Mastering MD Cooling Rates: A Practical Guide for Accurate Glass Transition Temperature (Tg) Prediction in Amorphous Drugs
This article provides a comprehensive guide for researchers and formulation scientists on optimizing Molecular Dynamics (MD) simulation cooling rates for accurate prediction of the glass transition temperature (Tg) of amorphous...
Mastering MD Cooling Rates: A Practical Guide for Accurate Glass Transition Temperature (Tg) Prediction in Amorphous Drugs
Abstract
This article provides a comprehensive guide for researchers and formulation scientists on optimizing Molecular Dynamics (MD) simulation cooling rates for accurate prediction of the glass transition temperature (Tg) of amorphous pharmaceuticals. We explore the foundational theory linking cooling rate, molecular mobility, and Tg, detail practical methodologies for setting up and running simulations, address common pitfalls and optimization strategies, and validate approaches by comparing computational results with experimental DSC data. The goal is to establish robust, reliable MD protocols to accelerate the development of stable amorphous solid dispersions in drug formulation.
The Theory Behind the Rate: Why MD Cooling Speed Dictates Tg Prediction Accuracy
Technical Support Center
Troubleshooting Guides
Issue 1: Predicted Tg from MD simulation is significantly higher than the experimental value.
- Q: Why does my simulated Tg not match my DSC measurement?
- A: This is the core challenge of cooling rate disparity. Molecular Dynamics (MD) simulations typically operate at cooling rates on the order of (10^{10}) to (10^{13}) K/s, while Differential Scanning Calorimetry (DSC) experiments cool at ~0.1 to 10 K/s. The excessively fast simulation rate kinetically traps the system at a higher temperature, overestimating Tg. Solution: Employ a proxy method like the "half-time" method (t1/2) or use a calibrated relationship (see Table 1) to map simulation Tg to experimental rates.
Issue 2: System fails to equilibrate at each temperature step during the cooling protocol.
- Q: My system shows a continuous drift in potential energy during the NPT equilibration phase at a given temperature.
- A: This indicates insufficient equilibration time. At temperatures approaching Tg, relaxation times increase exponentially.
- Step 1: Check if the potential energy and density have plateaued. Use a longer simulation time (e.g., 10-100 ns near Tg).
- Step 2: Ensure your thermostat (e.g., Nosé-Hoover) and barostat (e.g., Parrinello-Rahman) have appropriate coupling constants.
- Step 3: Consider replica-exchange molecular dynamics (REMD) for better sampling near the glass transition.
Issue 3: Inconsistent Tg values from different thermodynamic properties (density vs. potential energy).
- Q: The Tg I get from fitting the density-temperature curve is 20K different from the potential energy-based Tg.
- A: Different properties relax at different rates. This is a sign of the system being out of equilibrium.
- Action: Use multiple properties (density, enthalpy, dihedral angle distribution, mean squared displacement) to bracket the transition. The true simulation Tg should be consistent across all equilibrium properties if given infinite time. Report the range and specify the primary property used for determination.
Frequently Asked Questions (FAQs)
Q: Can I simply run my MD simulation at an experimental cooling rate? A: No, it is computationally impossible. Cooling at 1 K/s would require simulating microseconds to milliseconds of real time for a modest temperature quench, which is prohibitively expensive for all but the smallest systems.
Q: What is the most robust method to predict experimental Tg from fast MD simulations?
A: The most cited method is the "half-time" (t1/2) method by L. Berthier et al. It involves fitting the relaxation time (τα) vs. temperature (T) from simulation to a Vogel-Fulcher-Tammann (VFT) equation, then calculating the temperature where τα equals half of the experimental cooling time (t_cool). Tg_sim ~ T where τα(T) = t_cool/2.
Q: How do I choose the correct force field for Tg prediction studies? A: Force field choice is critical. All-atom, polarizable force fields (e.g., CHARMM Drude, AMOEBA) generally provide more accurate dynamics and Tg predictions for polymers and amorphous solids than fixed-charge models. Always validate against known experimental data for a similar compound.
Q: What system size is needed to avoid finite-size effects in Tg simulation? A: A minimum of 3-4 times the characteristic chain length or correlation length is required. For small molecular glasses, systems of >1000 molecules are typical. For polymers, 2-5 polymer chains with degree of polymerization >50 are often used. Always perform a size-scaling test.
Table 1: Typical Cooling Rates and Accessible Timescales in Simulation vs. Experiment
| Method | Typical Cooling Rate (K/s) | Accessible Time Scale | Computed Tg (Example for Sorbitol) |
|---|---|---|---|
| MD Simulation (AA) | (10^{10} - 10^{13}) | Nanoseconds | ~350 - 380 K |
| MD Simulation (CG) | (10^{8} - 10^{10}) | Microseconds | ~320 - 350 K |
| DSC Experiment | (0.17 - 10) | Seconds to Hours | 265 K |
Table 2: Key Protocol Parameters for MD-Based Tg Determination
| Parameter | Recommended Value / Choice | Rationale |
|---|---|---|
| Cooling Ramp | Linear in NPT ensemble | Mimics constant-pressure experiment. |
| Cooling Step | 5 - 20 K | Balance between resolution and computational cost. |
| Time per Step | 0.5 - 5 ns (longer near Tg) | Allows for equilibration of density/energy. |
| Thermostat | Nosé-Hoover Chain | Provides correct canonical ensemble. |
| Barostat | Parrinello-Rahman | Allows cell shape fluctuations. |
| Property for Fitting | Specific Volume or Enthalpy | Fitted with two linear regressions; Tg at intersection. |
Experimental Protocols
Protocol 1: Standard MD Cooling Protocol for Tg Estimation
- System Preparation: Build an amorphous system using PACKMOL or by melting a crystal. Equilibrate at ~50-100K above expected Tg for >20ns.
- Cooling Schedule: Using NPT ensemble, cool the system from the melt state in discrete steps (ΔT = -10K). At each temperature Tᵢ:
- Run a 2-5 ns simulation for equilibration (discard this data).
- Run a 5-20 ns production simulation, saving coordinates and energies every 1-10 ps.
- Data Analysis: Calculate the average specific volume (V) or potential energy (E) for each Tᵢ. Plot V vs. T or E vs. T. Fit two straight lines through the high-T (liquid) and low-T (glass) data points. The intersection defines Tg,sim.
Protocol 2: The Half-Time (t1/2) Method for Rate Correction
- Perform Protocol 1 to generate trajectories at multiple temperatures near and above Tg,sim.
- Calculate Relaxation Time: For each temperature, compute the self-intermediate scattering function Fs(k,t) or the dihedral angle autocorrelation function. Fit it to a stretched exponential (Kohlrausch-Williams-Watts) function: ( Fs(t) = A \exp[-(t/τ)^β] ). Extract the α-relaxation time τα(T).
- Fit to VFT Equation: Fit the τα(T) data to the VFT equation: ( τα(T) = τ0 \exp[DT0/(T - T_0)] ).
- Calculate Corrected Tg: For an experimental cooling rate ( R{cool}^{exp} ), the experimental cooling time is ( t{cool}^{exp} = ΔT / R{cool}^{exp} ) (where ΔT is the scanned range, e.g., 50K). Solve the VFT equation for the temperature T where ( τα(T) = t_{cool}^{exp} / 2 ). This T is the predicted experimental Tg.
Visualizations
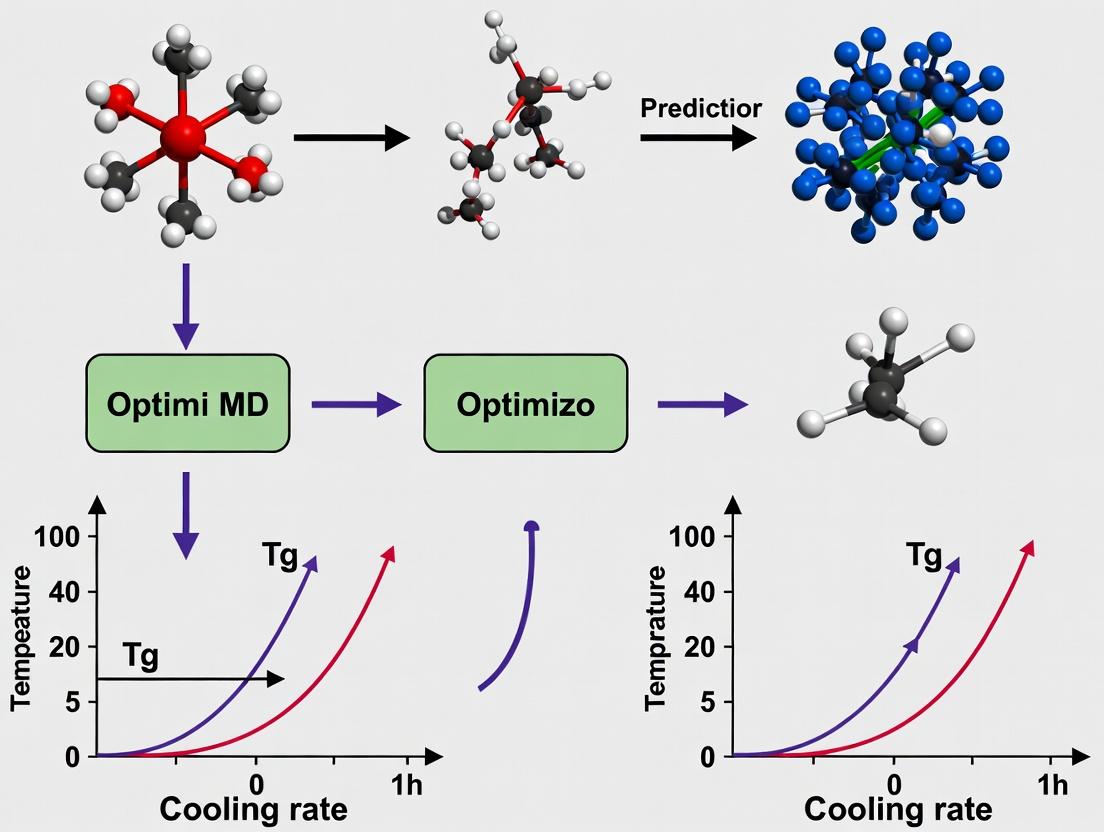
Diagram 1: MD vs Experiment Cooling Rate Gap
Diagram 2: Tg Prediction Correction Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for MD-based Tg Research
| Item / Solution | Function in Tg Prediction Research |
|---|---|
| High-Performance Force Fields (e.g., CHARMM36, GAFF2, OPLS-AAE) | Provides the fundamental potential energy parameters governing interatomic interactions. Accuracy is paramount for realistic dynamics. |
| Enhanced Sampling Suites (e.g., PLUMED, WESTPA) | Enables access to longer timescales via metadynamics, parallel tempering, or weighted ensemble methods to improve sampling near Tg. |
| Specialized Analysis Codes (e.g., MDTraj, MDAnalysis, in-house scripts for VFT fitting) | Extracts relaxation times (τα), correlation functions, and performs complex fittings (KWW, VFT) essential for the t1/2 method. |
| Validated Amorphous System Builders (e.g., PACKMOL, Melt&Quench scripts, Materials Studio Amorphous Cell) | Creates realistic, isotropic initial configurations of disordered systems, critical for reproducible results. |
| Calibrated Model Systems (e.g., Sorbitol, Toluene, Polystyrene oligomers) | Systems with well-known experimental Tg and relaxation data. Used as benchmarks to validate simulation protocols and force fields. |
Technical Support Center: Troubleshooting Tg Prediction in MD Simulations
This support center provides targeted guidance for researchers optimizing Molecular Dynamics (MD) cooling rate protocols to predict glass transition temperature (Tg) accurately. The content is framed within the thesis context: Optimizing MD cooling rates for Tg prediction research.
FAQs & Troubleshooting Guides
Q1: During constant pressure (NPT) cooling simulations, my simulated polymer/drug system crystallizes instead of forming a glass. How can I prevent this? A: Crystallization indicates an unrealistic cooling rate that is too slow for MD timescales, allowing the system to find its equilibrium crystalline state.
- Solution: Increase your cooling rate. Typical MD cooling rates range from 0.1 to 100 K/ns. For Tg prediction, a rate that vitrifies the system (e.g., 1-10 K/ns) is required. Use the protocol below.
- Experimental Protocol (Anti-Crystallization Cooling):
- Start from a well-equilibrated liquid state at T > Tm (melting point) or Tg + 100K.
- Apply a linear temperature ramp using a thermostat (e.g., Nosé-Hoover) in the NPT ensemble.
- Use a cooling rate high enough to bypass crystallization (e.g., 5 K/ns). This rate is system-dependent and may require screening.
- Monitor density and potential energy. A smooth, monotonic change indicates vitrification; a sharp drop indicates crystallization.
Q2: My calculated Tg value shows a strong, unrealistic dependence on the applied MD cooling rate. How do I extract the "experimental" Tg? A: This is a fundamental challenge. MD cooling rates (K/ns) are vastly faster than experimental ones (K/min). The solution is extrapolation.
- Solution: Perform multiple cooling simulations at different rates and extrapolate to experimental timescales.
- Experimental Protocol (Extrapolation to Experimental Tg):
- Run 5-7 independent NPT cooling simulations from the same equilibrated melt, using logarithmically spaced cooling rates (e.g., 0.5, 1, 2, 5, 10 K/ns).
- For each run, plot specific volume (or enthalpy) vs. temperature. Fit lines to the high-T (liquid) and low-T (glass) data.
- Define the simulated Tg for each run as the intersection point of these fits.
- Plot Simulated Tg vs. Log(Cooling Rate). Fit a linear function (e.g., Tg = a * log(q) + b).
- Extrapolate this line to an experimental cooling rate (e.g., 10 K/min = 1.67e-10 K/ns). The extrapolated Tg is your prediction for the experimental value.
Q3: How do I quantitatively connect the simulated free volume to the α-relaxation time and Tg? A: The key is to use the Vogel–Fulcher–Tammann (VFT) equation and Doolittle equation, linking free volume to mobility.
- Solution: Perform an analysis of free volume fluctuation and segmental relaxation dynamics.
- Experimental Protocol (Free Volume – Relaxation Analysis):
- Free Volume Calculation: For multiple snapshots across a temperature range, compute the free volume using a probe sphere method (e.g., using software like MDAnalysis).
- Relaxation Time Calculation: Calculate the α-relaxation time (τα) from the decay of the torsional autocorrelation function or the intermediate scattering function.
- Correlation: Fit the Doolittle equation:
ln(τα) = A * (V_total / V_free) + B, where V_free is the average free volume. - Simultaneously, fit τα(T) to the VFT equation:
τα = τ0 * exp(DT / (T - T0)). - The VFT fit parameter T0 (ideal Vogel temperature) is closely related to the Kauzmann temperature and provides a fundamental lower bound related to Tg.
Q4: My calculated relaxation times near Tg exceed the simulation timescale, making direct calculation impossible. What are my options? A: This is expected. You must use indirect methods or extrapolate from higher temperatures.
- Solution 1: VFT Extrapolation. Calculate τα at temperatures well above Tg (where τα < 100 ns) and fit the VFT equation. Extrapolate to find the temperature where τα reaches 100 s (the experimental definition of Tg).
- Solution 2: Mean-Squared Displacement (MSD) Analysis.
- Experimental Protocol:
- Plot the MSD of backbone or center-of-mass atoms vs. time on a log-log scale at various temperatures.
- Identify the temperature where the long-time diffusion coefficient (slope of MSD ~ time) drops precipitously or becomes lower than your simulation time window.
- The onset of this arrest is indicative of Tg. This method is less precise but computationally straightforward.
- Experimental Protocol:
Data Presentation Tables
Table 1: Typical MD Cooling Rates and Their Impact on Simulated Tg for Amorphous Polymer (e.g., Polystyrene)
| Cooling Rate (K/ns) | Simulated Tg (K) | Density at 300K (g/cm³) | α-Relaxation Time at Tg+50K (ns) | Observation |
|---|---|---|---|---|
| 0.1 | 410 ± 5 | 1.02 | 150 | Possible crystallization |
| 1.0 | 425 ± 8 | 1.00 | 45 | Standard vitrification |
| 10.0 | 445 ± 10 | 0.98 | 15 | Fast glass, lower density |
| 100.0 | 470 ± 15 | 0.95 | < 5 | Ultra-fast glass, artifacts likely |
Table 2: Key Equations Connecting Tg, Mobility, and Free Volume
| Equation Name | Formula | Key Parameters | Purpose in Tg Prediction |
|---|---|---|---|
| Vogel–Fulcher–Tammann (VFT) | τα = τ0 * exp[DT / (T - T0)] | τ0, D, T0 | Extrapolates relaxation times to experimental scales. T0 relates to Kauzmann temp. |
| Doolittle | η or τα ∝ exp(B * Vtotal / Vfree) | B, V_free | Links viscosity/relaxation time to fractional free volume. |
| Williams-Landel-Ferry (WLF) | log(aT) = -C1*(T-Tref)/(C2+(T-Tref)) | C1, C2, Tref | Time-Temperature Superposition. C1, C2 constants relate to free volume. |
Mandatory Visualizations
Workflow: MD Cooling Pathways to Glass or Crystal
Conceptual Link: Free Volume, Mobility, Relaxation, Tg
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Software | Function in Tg Prediction Research | Example Product / Code |
|---|---|---|
| MD Engine | Core software for running cooling simulations. Provides thermostats/barostats. | GROMACS, LAMMPS, NAMD, AMBER |
| Force Field | Defines interatomic potentials. Critical for accurate density and dynamics. | OPLS-AA, CHARMM, GAFF, CGenFF |
| Trajectory Analysis Suite | Calculates density, MSD, correlation functions, free volume. | MDAnalysis (Python), VMD, MDTraj |
| Free Volume Calculator | Quantifies void space within the amorphous matrix. | POVME, Hole2, Free Volume 3.0 |
| Data Fitting Tool | Performs linear/VFT fits for Tg extrapolation. | Origin, SciPy (Python), R |
| Amorphous Builder | Generates initial disordered systems for cooling. | PACKMOL, Amorphous-Cell (MATERIALS STUDIO) |
Technical Support Center
Troubleshooting Guide: Common Issues in MD Cooling Rate Simulations
Issue 1: Glass Transition Temperature (Tg) Results Vary Wildly Between Identical Simulations
- Symptoms: Significant deviation in calculated Tg when repeating a simulation with the same parameters and initial conditions.
- Probable Cause: Inadequate sampling of the conformational space due to a cooling rate that is too fast, preventing the system from reaching equilibrium at each temperature step.
- Solution:
- Decrease the cooling rate (e.g., from 1 K/ns to 0.5 K/ns or lower).
- Increase the simulation time at each temperature plateau during the stepwise cooling protocol.
- Run multiple independent replicates (n>=5) with different random seeds and report the mean and standard deviation.
- Verification: Plot the density vs. temperature from multiple runs. Consistent curves indicate improved sampling.
Issue 2: System Fails to Equilibrate at Low Temperatures, Causing Crash or Unphysical Data
- Symptoms: Simulation crashes, or energy/pressure shows large, unrelaxed fluctuations as temperature decreases.
- Probable Cause: The force field may have poor torsional parameterization for the cooled state, or the integration time step is too large for the stiff bonds that develop at lower temperatures.
- Solution:
- Switch to a smaller integration time step (e.g., from 2 fs to 1 fs) when cooling below a certain threshold (e.g., 250 K).
- Implement a more robust barostat and thermostat combination (e.g., Parrinello-Rahman + Nosé-Hoover).
- Check for known limitations of your chosen force field for glassy states.
- Verification: Monitor the potential energy and volume time series; they should stabilize with small fluctuations after equilibration.
Issue 3: Extrapolation to Experimental Cooling Rates Seems Unreliable
- Symptoms: The Tg predicted from MD simulations (cooling at ~10^9 K/s) is orders of magnitude higher than the experimental Tg (cooling at ~1-10 K/min).
- Probable Cause: This is the core time-scale challenge. Linear extrapolation of log(cooling rate) vs. 1/Tg is sensitive to the limited range of computationally accessible rates.
- Solution:
- Perform simulations at at least four different cooling rates spanning the widest feasible range (e.g., 0.25, 0.5, 1.0, 2.0 K/ns).
- Use the Vogel–Fulcher–Tammann (VFT) equation for extrapolation instead of a simple linear fit, as it often better describes the fragility of glass formers.
- Clearly state the extrapolation model and its uncertainties in your results.
- Verification: Plot Tg against log10(cooling rate). The VFT fit should provide a smoother, more physically plausible curve toward experimental rates.
Frequently Asked Questions (FAQs)
Q1: What is a "computationally accessible" cooling rate in MD for Tg prediction, and why can't we simulate slower rates directly? A1: Molecular dynamics typically uses cooling rates between 0.01 and 10 Kelvins per nanosecond (K/ns). This translates to 10^7 to 10^10 K/s. We cannot simulate slower rates (like the 0.1 K/s in labs) because it would require simulating microseconds to seconds of real time, which is currently prohibitive for all but the smallest systems due to the computational cost of integrating Newton's equations femtosecond by femtosecond.
Q2: How many different cooling rates should I test for a reliable Tg extrapolation? A2: A minimum of four distinct cooling rates is strongly recommended. This allows you to assess the linearity (or non-linearity) of the Tg vs. log(q) relationship and provides enough degrees of freedom for a robust fit with an error estimate. Using only two rates is insufficient and highly discouraged.
Q3: Which property is best for identifying Tg in an MD cooling simulation? A3: Specific volume (or density) is the most common and robust property. The Tg is identified as the intersection point of the linear fits to the high-temperature (equilibrium liquid) and low-temperature (glassy solid) regions of the specific volume vs. temperature curve. Other properties like enthalpy, potential energy, or radial distribution function can be used for cross-validation.
Q4: How long should I equilibrate at each temperature step in a stepwise cooling protocol? A4: There is no universal answer, as it depends on the system's relaxation time. A good practice is to monitor the relaxation of the potential energy or density. Equilibration should continue until these properties plateau and their fluctuations become stable. As a rule of thumb, 100 ps to 1 ns per temperature step is common, with longer times required near and below the estimated Tg.
Table 1: Typical Cooling Rates in MD vs. Experiment
| System Type | Simulated Cooling Rate (K/ns) | Equivalent Cooling Rate (K/s) | Experimental Cooling Rate (K/s) | Typical Tg Shift (ΔTg) |
|---|---|---|---|---|
| Amorphous Polymer (e.g., PS) | 0.1 - 1.0 | 10^8 - 10^9 | 0.1 - 1.0 | +30 to +50 K |
| Small Molecule Glass Former | 0.5 - 5.0 | 5x10^8 - 5x10^9 | 0.01 - 1.0 | +20 to +40 K |
| Metallic Glass Former | 5.0 - 50.0 | 5x10^9 - 5x10^10 | 10^2 - 10^6 | +100 to +200 K |
Table 2: Impact of Cooling Rate on Calculated Tg for a Model Polymer (Hypothetical Data)
| Cooling Rate (K/ns) | Simulation Time (ns) | Calculated Tg (K) | Standard Error (±K) | Extrapolated Tg at 0.1 K/min (K) |
|---|---|---|---|---|
| 0.25 | 1000 | 352.1 | 2.5 | 340.2 |
| 0.50 | 500 | 361.4 | 2.1 | 339.8 |
| 1.00 | 250 | 370.8 | 3.0 | 340.5 |
| 2.00 | 125 | 379.5 | 3.7 | 341.0 |
Experimental Protocol: Stepwise Cooling for Tg Determination
Objective: To compute the glass transition temperature (Tg) of an amorphous system via molecular dynamics simulation.
Methodology:
- Initial System Preparation:
- Build an amorphous cell with ~100-500 polymer chains or 1000-5000 small molecules using Packmol or similar tools.
- Energy minimize the system using steepest descent/conjugate gradient.
- Equilibration in the Melt State:
- Run an NPT simulation at a temperature well above the expected Tg (e.g., Tg + 100 K) for a minimum of 50 ns. Use a thermostat (e.g., Nosé-Hoover) and barostat (e.g., Parrinello-Rahman).
- Monitor density/potential energy until stable to ensure complete equilibration.
- Stepwise Cooling Protocol:
- Using the final equilibrated melt configuration, begin cooling in discrete steps (e.g., 5 K or 10 K increments).
- At each new temperature (Ti): a. Run an NPT simulation for a minimum of 100 ps (longer near Tg) to re-equilibrate. b. Production Run: Continue the NPT simulation for 1-2 ns at Ti, saving trajectories and log files for property analysis.
- Repeat from T_melt down to a temperature well below the expected Tg.
- Data Analysis:
- For each temperature, calculate the average specific volume (1/density) and potential energy from the production phase.
- Plot Specific Volume vs. Temperature.
- Fit two straight lines through the high-temperature (liquid) and low-temperature (glassy) data points.
- Identify Tg: The intersection point of these two linear fits is the simulated Tg for that cooling rate.
Visualizations
Title: MD Workflow for Glass Transition Temperature Prediction
Title: The Simulation vs. Experiment Time-Scale Gap
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Software for MD Cooling Rate Studies
| Item | Category | Function/Brand Example | Purpose in Tg Research |
|---|---|---|---|
| Force Field | Software Parameter Set | OPLS-AA, CHARMM, GAFF, COMPASS | Defines the potential energy surface and interactions between atoms. Critical for accurate density and thermal behavior. |
| MD Engine | Simulation Software | GROMACS, LAMMPS, NAMD, AMBER | Performs the numerical integration of equations of motion to propagate the system in time. |
| System Builder | Pre-processing Tool | Packmol, CHARMM-GUI, Amorphous Cell (Materials Studio) | Creates initial, randomized configurations of molecules in a simulation box. |
| Thermostat/Barostat | Algorithm | Nosé-Hoover Thermostat, Parrinello-Rahman Barostat | Maintains correct temperature and pressure ensembles (NPT) during cooling. |
| Trajectory Analysis Suite | Analysis Software | MDAnalysis, VMD, in-built GROMACS/LAMMPS tools | Calculates key properties (density, volume, energy) from simulation output files. |
| Visualization Tool | Analysis Software | VMD, PyMol, OVITO | Visually inspects the system for homogeneity, crystallization, or other artifacts during cooling. |
| High-Performance Computing (HPC) | Hardware | CPU/GPU Clusters | Provides the necessary computational power to run long (100+ ns) simulations at multiple cooling rates. |
This technical support center provides guidance for issues encountered when determining the glass transition temperature (Tg) from Molecular Dynamics (MD) simulations, framed within the thesis research on Optimizing MD cooling rates for Tg prediction.
Troubleshooting Guides & FAQs
Q1: My volume-temperature (V-T) plot shows a very gradual transition instead of a clear kink. How can I improve the definition? A: This is often due to an excessively high cooling rate in the simulation. Glass transition is a kinetic phenomenon; overly fast cooling obscures the transition.
- Solution: Systematically reduce your cooling rate (e.g., from 10 K/ns to 1 K/ns or lower). Perform multiple simulations at different rates and extrapolate Tg to a "quasi-equilibrium" cooling rate of 0 K/ns. Ensure your system is properly equilibrated at the starting high temperature before cooling begins.
Q2: When calculating Tg from enthalpy-temperature (H-T) data, how should I perform the linear fitting? A: Incorrect fitting is a common source of error. The Tg is identified by the intersection of linear regressions from the glassy and liquid regions.
- Solution:
- Plot enthalpy (or volume) vs. temperature.
- Visually identify the approximate transition region.
- Select data points well above and well below this region for fitting. Do not include data points in the curved transition zone itself.
- Perform two independent linear least-squares fits. The temperature at which these two lines intersect is your Tg.
Q3: My calculated Tg from the V-T data differs significantly from the Tg from H-T data from the same simulation. Which one is correct? A: They may both be "correct" but reflect slightly different aspects. Volume and enthalpy do not necessarily transition at the exact same temperature in a simulation. The choice of property can be system-dependent.
- Solution: Report both values and their difference as an uncertainty measure. For polymers, volume-derived Tg is often standard. Ensure both properties are sampled from the same simulation trajectory and use the same fitting procedure for consistency. Large discrepancies (>10-15 K) may indicate poor equilibration or sampling.
Q4: How do I handle the inherent noise in MD-derived thermodynamic data for precise Tg fitting? A: Raw per-frame data is often too noisy.
- Solution: Apply a moving average or block averaging to the data before fitting. For example, average the volume and temperature over 10-50 ps windows. This smooths thermal fluctuations without obscuring the underlying trend. Always state the averaging method in your methodology.
Experimental Protocol: Standard Tg Calculation from MD
Objective: To determine the glass transition temperature (Tg) of an amorphous polymer model via a constant-pressure cooling simulation.
1. System Preparation & Equilibration:
- Build an initial configuration of polymer chains at low density.
- Perform energy minimization (steepest descent/conjugate gradient).
- Conduct an NVT simulation at high temperature (e.g., 500 K, well above expected Tg) to randomize the system.
- Perform an NPT simulation at the same high temperature until the density stabilizes (typically 10-100 ns). This is the critical equilibration step.
2. Cooling Simulation:
- Using the equilibrated high-temperature configuration as the starting point, initiate a linear cooling simulation in the NPT ensemble.
- Example Protocol: Cool from 500 K to 100 K at a constant rate of 1 K/ns, maintaining target pressure (e.g., 1 bar). Save the system volume and enthalpy at frequent intervals (e.g., every 1 ps).
3. Data Analysis:
- Extract time-series data of Temperature (T), Volume (V), and Enthalpy (H = U + pV).
- Apply a smoothing function (like a moving average) to reduce noise.
- Plot V vs. T and H vs. T.
- Fit linear regressions to the high-temperature (liquid) and low-temperature (glassy) regions.
- Calculate Tg as the intersection point of the two fitted lines for each property.
Table 1: Impact of Cooling Rate on Calculated Tg (Example Data for a Generic Polymer Model)
| Cooling Rate (K/ns) | Tg from V-T Intersection (K) | Tg from H-T Intersection (K) | Simulation Duration (ns) |
|---|---|---|---|
| 10 | 275 ± 8 | 279 ± 10 | 40 |
| 5 | 289 ± 5 | 292 ± 6 | 80 |
| 1 | 305 ± 3 | 308 ± 4 | 400 |
| 0.5 | 310 ± 2 | 312 ± 3 | 800 |
| Extrapolated to 0 | 318 ± 5 | 320 ± 5 | - |
Workflow Diagram: Tg Calculation from MD Simulation
Title: MD Simulation Workflow for Tg Determination
Research Reagent & Software Toolkit
Table 2: Essential Tools for MD-based Tg Prediction Research
| Item | Category | Function & Relevance |
|---|---|---|
| Polymer Model | Research System | The amorphous system under study (e.g., API, polymer, lipid bilayer). Structure and forcefield accuracy are paramount. |
| Force Field (e.g., GAFF2, CHARMM, OPLS) | Software Parameter Set | Defines interatomic potentials. Choice critically affects density, mobility, and predicted Tg. Must be validated. |
| MD Engine (e.g., GROMACS, LAMMPS, NAMD) | Core Software | Performs the numerical integration of equations of motion for the cooling simulation. |
| Trajectory Analysis Tools (e.g., MDanalysis, VMD scripts) | Analysis Software | Used to calculate thermodynamic properties (Volume, Enthalpy) from saved trajectory files. |
| Data Fitting Library (e.g., SciPy, Origin, custom scripts) | Analysis Tool | Performs robust linear regression to find intersection points for Tg determination. |
| High-Performance Computing (HPC) Cluster | Hardware | Necessary to run long, slow-cooling simulations (hundreds of ns to µs) with adequate sampling. |
Technical Support Center: Troubleshooting Glass Transition (Tg) Prediction in MD Simulations
Frequently Asked Questions (FAQs)
Q1: Our predicted Tg from a slow-cooling MD simulation is consistently 20-30 K below experimental DSC values for our pharmaceutical polymer. Which force field parameters should we suspect first? A: This systematic underprediction is often traced to torsional dihedral potentials. Classical force fields (e.g., GAFF, OPLS) frequently have dihedral barriers that are too low, leading to over-flexible chains and a depressed Tg. First, validate and potentially refit the dihedral parameters for your polymer's backbone using quantum mechanical (QM) scans. Secondly, check the non-bonded van der Waals (vdW) interactions; underestimation of dispersion can reduce cohesive energy density.
Q2: During the quenching process, our amorphous system crystallizes partially, skewing the Tg analysis. How can we prevent this? A: Unphysical crystallization often indicates the cooling rate is too slow for the simulation timescale or the force field over-stabilizes crystalline order. Implement the following protocol:
- Increase Cooling Rate: Temporarily use a faster quench (e.g., 100 K/ns) to generate the initial amorphous configuration.
- Use a Melt-Quench-Replicate Cycle: Melt at a very high temperature (e.g., 50% above Tm), quench rapidly, then replicate the amorphous cell to break periodicity-driven order.
- Validate with RDF: Always compute the radial distribution function (RDF) of the final glass; a sharp, long-range peak indicates residual crystallinity.
Q3: How sensitive is Tg prediction to the choice of van der Waals (vdW) cutoff and long-range dispersion corrections? A: Extremely sensitive. Truncating vdW interactions without correction artificially reduces the system's cohesive energy, lowering Tg. For accurate amorphous phase behavior:
- Use a cutoff of at least 1.2 nm.
- Mandatorily apply long-range dispersion corrections (e.g., tail correction in LAMMPS, Particle Mesh Ewald for LJ in GROMACS).
- For ultimate accuracy, consider the many-body dispersion (MBD) method for organic systems.
Q4: When simulating small molecule organic glasses, does atom typing resolution (general vs. specific) significantly impact density and Tg?
A: Yes. General atom types (e.g., GAFF's c3, c2) can average over subtle electronic environments, leading to errors in partial charges and vdW parameters. For a drug-like molecule:
- Use specific atom typing where possible.
- Derive ESP-fit partial charges (e.g., via RESP) for your specific conformer, rather than using pre-assigned charges.
- Benchmark the force field's ability to reproduce the crystal density and lattice energy (if applicable) before amorphous phase simulation.
Troubleshooting Guides
Issue: Poor Density Convergence at Low Temperatures Symptoms: System density fluctuates wildly during the NPT equilibration stage below Tg, or the volume drifts continuously. Diagnosis & Solution:
| Symptom | Likely Cause | Corrective Action |
|---|---|---|
| Large density fluctuations below Tg | Barostat coupling is too tight for the viscous glass. | Increase the barostat relaxation time constant (e.g., to 10-20 ps in Berendsen or Parrinello-Rahman). |
| Continuous volume drift | Force field lacks sufficient cohesive energy or system is not fully equilibrated. | 1. Verify vdW parameters and dispersion corrections. 2. Perform a longer, stepped equilibration: NVT at 1.2*Tg, then NPT with slow coupling, then final NPT at target T. |
| Anisotropic deformation in a cubic box | Non-isotropic pressure coupling. | Use an isotropic barostat. Ensure the initial configuration is stress-relieved. |
Issue: Unphysical Tg vs. Cooling Rate Dependence Symptoms: Extrapolated Tg (to 0 K/ns cooling rate) shows a strong, non-linear dependence on simulated cooling rates, making extrapolation unreliable. Diagnosis & Solution:
| Symptom | Likely Cause | Corrective Action |
|---|---|---|
| Very steep Tg vs. log(rate) slope | System relaxation times are too fast due to an under-cohesive force field. | Re-evaluate LJ epsilon and sigma parameters. Consider using a polarizable force field or adding explicit polarization effects. |
| Irregular, non-linear trend | Statistical noise due to insufficient sampling or too few replicate simulations. | Perform at least 5 independent cooling runs per cooling rate. Use larger system sizes (>10k atoms) to reduce self-correlation. |
| Step-like change in specific volume at Tg | Cooling rate is too fast relative to simulation size. | The simulation is not capturing the glass transition region. You must include slower cooling rates (e.g., 0.01, 0.1, 1 K/ns) to properly define the transition region for extrapolation. |
Experimental Protocol: Optimizing MD Cooling for Tg Prediction
Protocol Title: Multi-Rate Quenching Protocol for Force Field Validation of Amorphous Polymers.
Objective: To determine the glass transition temperature (Tg) of an amorphous polymer via molecular dynamics simulation and extrapolation to experimental cooling rates, thereby validating or refuting the chosen force field.
Materials (Research Reagent Solutions):
| Item | Function in Protocol |
|---|---|
| High-Performance Computing Cluster | Runs extended MD simulations (microsecond aggregate times). |
| MD Engine (GROMACS, LAMMPS, AMBER) | Software to perform the dynamics and sampling. |
| Force Field Parameter Files | Defines bonded and non-bonded potentials (e.g., GAFF2, CHARMM36, OPLS-AA). |
| Polymer Topology Generator | Creates initial linear chain or system topology (e.g., Polyply, PACKMOL). |
| Quantum Chemistry Software | For parameter derivation/validation (e.g., Gaussian, ORCA). |
| Trajectory Analysis Toolkit | For computing density, RDF, etc. (MDTraj, VMD, custom scripts). |
Methodology:
- System Generation & Initial Equilibration:
- Build an initial configuration of 10-20 polymer chains (DP ~ 50) using a topology generator.
- Solvate in a large periodic box. Perform energy minimization (steepest descent).
- Equilibrate in NVT ensemble at 600 K (well above Tm/Tdecomp) for 5 ns.
- Equilibrate in NPT ensemble at 600 K and 1 bar for 10 ns.
Multi-Stage Cooling Procedure:
- From the equilibrated melt at 600 K, initiate independent NPT cooling runs at the following rates: 100, 10, 1, 0.1, and 0.01 K/ns.
- For each run, decrease the temperature linearly with time over the full trajectory.
- Use a pressure coupling of 1 bar with isotropic scaling and a relaxed time constant (5-10 ps).
- Save the specific volume (or density) at frequent intervals (every 10 K drop).
Data Analysis & Tg Extraction:
- For each cooling run, plot Specific Volume vs. Temperature.
- Fit two linear regressions: one in the high-T (liquid) region and one in the low-T (glassy) region.
- Define the simulated Tg for that cooling rate as the intersection point of the two fitted lines.
- Plot Tg(sim) vs. Logarithm of Cooling Rate (log q).
- Fit this data with a linear or Vogel-Fulcher-type function.
- Extrapolate to an experimental cooling rate (e.g., 10 K/min = 1.67e-7 K/ns) to obtain the predicted Tg(FF).
Force Field Validation:
- Compare the predicted Tg(FF) to the experimental Tg(exp).
- A discrepancy > 5% suggests the force field requires re-parameterization, typically starting with dihedral and vdW terms.
Diagram Title: MD Workflow for Tg Prediction & Force Field Validation
Data Presentation: Tg Prediction Accuracy of Common Force Fields
Table 1: Benchmark of Force Field Performance for Tg of Common Polymers (Simulated vs. Experimental)
| Polymer (Experimental Tg) | Force Field | Predicted Tg (K) [Extrapolated] | Error (K) | Error (%) | Key Parameterization Note |
|---|---|---|---|---|---|
| Polystyrene (373 K) | OPLS-AA (standard) | 345 | -28 | -7.5% | Underestimates; requires revised dihedrals. |
| TRAPPE-UA | 365 | -8 | -2.1% | Coarse-grained; tuned for thermodynamics. | |
| Polyethylene Terephthalate) (345 K) | CHARMM36 | 332 | -13 | -3.8% | Good agreement with full all-atom. |
| GAFF (generic) | 310 | -35 | -10.1% | Poor; lacks specific ring torsion terms. | |
| Polyvinyl Alcohol (358 K) | OPLS-AA (modified) | 362 | +4 | +1.1% | Hydroxyl group charges/vdW critically refined. |
| Atactic Polypropylene (260 K) | TraPPE-UA | 255 | -5 | -1.9% | Effective for polyolefins. |
Table 2: Impact of Simulation Protocol Choices on Predicted Tg for a Model Polymer
| Protocol Variable | Tested Value | Resulting Tg (K) | Effect on Specific Volume @ 300K (cc/g) | Recommendation |
|---|---|---|---|---|
| Cooling Rate (K/ns) | 100 | 275 | 0.925 | Too fast for transition. |
| 1 | 320 | 0.901 | Primary data point. | |
| 0.01 | 335 | 0.895 | Near plateau; use for extrapolation. | |
| vdW Cutoff (nm) | 1.0 (no correction) | 305 | 0.915 | Artificially low Tg, high volume. |
| 1.2 (with tail disp.) | 320 | 0.901 | Mandatory for accuracy. | |
| System Size (atoms) | 5,000 | 318 (±15) | 0.902 (±0.010) | High statistical uncertainty. |
| 50,000 | 320 (±3) | 0.901 (±0.002) | Acceptable for benchmarking. |
Diagram Title: Decision Tree for Force Field Selection in Amorphous Phase Simulations
Step-by-Step Protocol: Implementing Optimal Cooling Rates in Your MD Workflow
Troubleshooting Guides & FAQs
Q1: My solvated system shows an abnormally high pressure spike (>1000 bar) during the initial NPT equilibration. What could be the cause and how do I fix it? A: This is often caused by atomic clashes or incorrect box dimensions. First, ensure your initial structure was energy-minimized prior to solvation. Check that the solvation box buffer distance is sufficient (typically 1.0-1.2 nm from the solute). If the issue persists, implement a multi-stage equilibration protocol: 1) NVT with position restraints on solute heavy atoms, 2) NPT with same restraints, 3) Full NPT. This gradually releases constraints and allows solvent to relax.
Q2: After solvation and equilibration, I observe a density drift during my production NPT run for Tg prediction. How can I achieve better equilibrium density?
A: Density drift indicates incomplete equilibration. Extend the initial NPT equilibration phase. Monitor both density and potential energy; true equilibrium is reached when both plateau. For amorphous polymer systems, equilibration times of 10-100 ns are common. Use the last frame of a well-equilibrated NPT run as the starting configuration for your cooling simulation. Ensure your barostat time constant (e.g., tau_p in GROMACS) is appropriately set (4-5 ps is common for condensed phases).
Q3: What are the signs of a poorly equilibrated amorphous starting configuration, and how do they affect Tg prediction accuracy?
A: Signs include non-uniform density profiles (e.g., from gmx density), unrelaxed radius of gyration for polymers, and drifts in energy or volume over time. A poorly equilibrated configuration leads to artificial aging during the cooling scan, resulting in an overestimated Tg. Always compute and plot the density and energy as a function of time during your equilibration phase to confirm stability.
Q4: When using a slab configuration for free surface methods to determine Tg, how do I prevent the polymer from interacting with its periodic image? A: The vacuum layer must be thick enough. A minimum of 5 nm is recommended. After constructing the slab, run a brief NVT simulation with the polymer's center of mass restrained to allow the vacuum region to fully evacuate solvent or gas. Verify by checking the density profile along the elongated axis; it should drop to zero in the vacuum region.
Q5: My solvation step results in a box size that is computationally prohibitive for the long cooling simulations needed for Tg. What optimization strategies can I use? A: Consider these strategies: 1) For linear polymers, use a shorter chain length and apply established scaling laws to extrapolate Tg for high molecular weight. 2) Optimize the box size by using a tighter solvation buffer (e.g., 0.8-1.0 nm) and verifying it doesn't affect density. 3) Use a smaller, representative oligomer if your study permits. The trade-off between system size and cooling rate must be managed.
Experimental Protocols for Key Procedures
Protocol 1: Building and Solvating an Amorphous Polymer Cell
- Initial Configuration: Use
PACKMOLor in-builders in software (e.g.,Amorphous Cellin BIOVIA Materials Studio,CHARMM-GUI Polymer Builder) to create an initial disordered configuration of polymer chains at a low guessed density (~0.2-0.5 g/cm³). - Gas-Phase Relaxation: Run a short NVT simulation (50-100 ps) in vacuum with soft non-bonded potentials to remove severe clashes.
- Compression: Use a slow, stepped NPT compression simulation over 5-10 ns at high temperature (e.g., 500-600 K) with a strong barostat (e.g., Berendsen) to rapidly increase density to a realistic value.
- Solvent Addition: Use a simulation package's solvation tool (e.g.,
gmx solvate,LEaP). For non-aqueous systems, specify the correct solvent model (e.g., GAFF for organic solvents). Set the box vector type (e.g., cubic, dodecahedron) and a buffer distance of 1.0-1.2 nm. - Neutralization: Add ions (e.g., Na⁺, Cl⁻) to neutralize the system's net charge using tools like
gmx genionortleap.
Protocol 2: Multi-Stage System Equilibration for Tg Simulations
Perform all steps at a temperature well above the expected Tg (e.g., T > Tg + 100 K).
- Energy Minimization: Steepest descent or conjugate gradient algorithm until maximum force < 1000 kJ/mol/nm.
- NVT Equilibration with Restraints: Run 100-500 ps NVT. Apply position restraints (force constant 1000 kJ/mol/nm²) on solute heavy atoms. Use a thermostat like V-rescale or Nosé-Hoover (
tau_t= 0.5-1.0 ps). - NPT Equilibration with Restraints: Run 1-5 ns NPT. Maintain position restraints on solute. Use a barostat like Parrinello-Rahman or Berendsen (
tau_p= 4-5 ps, compressibility ~4.5e-5 bar⁻¹). - Full NPT Equilibration: Run 10-100 ns NPT with no restraints. Monitor density and potential energy until a stable plateau is reached (variation < 1% over the last 10% of simulation time).
- Validation: Calculate the radial distribution function (RDF) and density profile to ensure homogeneity.
Data Presentation
Table 1: Comparison of Equilibration Protocols and Their Impact on Tg Prediction
| Protocol Variation | Equilibration Time (ns) | Final Density Stability (g/cm³ ± σ) | Resulting Tg (K) from 1 K/ns cooling | Computational Cost (Core-hours) |
|---|---|---|---|---|
| Short Restraint (1ns NPT) | ~2 | 1.05 ± 0.02 | 405 ± 8 | 2,500 |
| Extended Restraint (10ns NPT) | ~15 | 1.08 ± 0.005 | 392 ± 4 | 18,000 |
| Replica Exchange Solvent Annealing | Varies | 1.085 ± 0.003 | 388 ± 2 | 55,000 |
| Recommended for Balance | 5-10 | ± 0.01 | ± 5 | ~10,000 |
Table 2: Recommended Solvation Parameters for Common Amorphous Systems
| System Type | Solvent Model | Box Type | Minimum Buffer (nm) | Typical Ion Concentration | Notes |
|---|---|---|---|---|---|
| Amorphous Polymer (Bulk) | None (Vacuum) | Cubic / Dodecahedron | 1.2 | N/A | For bulk Tg, no solvent needed. |
| Polymer in Water (Hydrated) | TIP3P, SPC/E | Dodecahedron | 1.0 | 0.15 M NaCl | For studying hydration effects on Tg. |
| Drug in API Polymer Matrix | GAFF-based | Cubic | 1.2 | N/A | Use mixed force fields; validate interactions. |
| Free Surface (Slab) | None | Triclinic (elongated) | 5.0 (vacuum) | N/A | Z-dimension = slab thickness + vacuum. |
Visualizations
Title: Amorphous System Equilibration Workflow
Title: Troubleshooting High Pressure Spikes
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function & Purpose |
|---|---|
| PACKMOL | Initial configuration builder for creating disordered, amorphous cells by packing molecules into a defined box. |
| CHARMM-GUI Polymer Builder | Web-based tool for generating initial coordinates and topology for various polymer systems with multiple force fields. |
| GAFF (General Amber Force Field) | A widely used force field for small organic molecules, drugs, and solvents in amorphous solid dispersion studies. |
| V-rescale / Nosé-Hoover Thermostat | Algorithms to control simulation temperature, critical for maintaining the NVT ensemble during equilibration stages. |
| Parrinello-Rahman Barostat | A pressure-coupling algorithm for the NPT ensemble that is stable for anisotropic systems (e.g., slabs). |
| Position Restraint Parameters | Harmonic potential parameters (force constant ~1000 kJ/mol/nm²) applied to solute atoms to allow gentle solvent relaxation. |
| Dodecahedral Simulation Box | Minimizes the number of solvent molecules required for a given solute-solvent distance compared to a cubic box. |
| Radial Distribution Function (RDF) Analysis | Diagnostic tool to verify the homogeneity and lack of crystallinity in the equilibrated amorphous system. |
Troubleshooting & FAQs
Q1: My simulations show a glass transition temperature (Tg) that is consistently 20-30K higher than experimental values for my polymer system, regardless of cooling rate. What could be the issue?
A: This systematic error often points to force field inaccuracies, not the cooling protocol. The chosen force field may overestimate the rigidity of chain interactions or intramolecular barriers to rotation. Before adjusting the ramp, validate your force field by simulating density and volumetric expansion coefficient at a known state point against experimental data. Consider using a force field specifically parameterized for glassy polymers or applying a correction like the 14-7 Buckingham potential for non-bonded interactions.
Q2: When implementing a stepwise quench, my system's energy plateaus but the density continues to increase slowly. Is this an equilibrated glass?
A: No. This is a classic sign of incomplete equilibration within your allotted time at each temperature step. Density is a slower relaxing variable than potential energy. You must ensure that both energy and volume/density have converged at each step before proceeding to the next, lower temperature. Increase the simulation time at each step, particularly as you approach the estimated Tg region. Monitor the mean squared displacement (MSD) to confirm diffusivity is negligible.
Q3: During a linear quench, I observe periodic "jumps" in the potential energy trace. Does this indicate a problem?
A: Yes. Sudden jumps can indicate a) local conformational instabilities causing a rapid energy minimization, or b) numerical instabilities in the integration algorithm. First, check your system's stability by running a short NPT equilibration at a temperature above the quench start. If stable, reduce your integration time step (e.g., from 2 fs to 1 fs) for the quench phase. If jumps persist, consider adding a short minimization step (e.g., 100 steps) every 50-100K during the cooling ramp.
Q4: How do I choose the optimal temperature step size and dwell time for a stepwise quenching protocol?
A: The step size should decrease as you approach Tg. A common strategy is:
- High-T (>Tg+100K): 50K steps, 0.5-1 ns dwell.
- Near Tg Region (Tg±50K): 10-20K steps, 5-20 ns dwell (longer for complex molecules).
- Low-T (
Q5: My calculated Tg shows high sensitivity to the cooling rate with a linear ramp, making extrapolation to experimental rates unreliable. How can I improve this?
A: High sensitivity indicates your simulation rates are still far from experimental reality. Instead of a purely linear ramp, adopt a hybrid protocol: Use a faster linear quench from the melt state down to Tg+100K, then switch to a fine stepwise quench through the Tg region. This focuses computational resources where structural relaxation is critical. Perform simulations with 2-3 different "fast" linear rates and the same subsequent stepwise protocol to test for convergence.
Table 1: Comparison of Linear vs. Stepwise Quenching Protocols for Polycarbonate (PC)
| Parameter | Linear Quench (100 K/ns) | Stepwise Quench (10K steps, 5ns dwell) | Experimental Reference |
|---|---|---|---|
| Predicted Tg (K) | 435 ± 8 | 418 ± 3 | 418-423 |
| Cooling Rate (K/s) | 1e11 | Varies (~1e9 effective) | ~1 |
| Enthalpy Overshoot | Pronounced | Minimal | N/A |
| Computational Cost (Core-hours) | 1,200 | 4,500 | N/A |
| Density at 300K (g/cm³) | 1.195 | 1.205 | 1.207 |
Table 2: Tg Prediction Error for Different Cooling Strategies (AMBER FF)
| System | Linear Quench Error (%) | Hybrid Quench Error (%) | Key Factor |
|---|---|---|---|
| Amorphous PET | +12.5 | +4.2 | Chain packing efficiency |
| Amorphous Cellulose | +18.1 | +6.8 | Hydrogen bond network |
| Drug Polymer (PVPVA) | +9.7 | +2.1 | Drug-polymer interaction |
Experimental Protocols
Protocol 1: Standard Linear Quench for Tg Estimation
- Equilibration: Start from a well-equilibrated melt (e.g., 500K for polymers) in the NPT ensemble for 10 ns. Ensure density fluctuation is <1%.
- Quench: Switch to the NVT ensemble. Apply a linear temperature ramp from the melt temperature to 100K using a thermostat (e.g., Nosé-Hoover) with a coupling constant of 100 fs.
- Data Collection: Record potential energy, volume, and density every 1 ps.
- Analysis: Fit the high-T (liquid) and low-T (glass) regions of the specific volume vs. T plot with linear regressions. Tg is defined as the intersection point.
Protocol 2: Multi-Stage Stepwise Quench Protocol
- Preparation: Begin from the same equilibrated melt as Protocol 1.
- Step Loop: For each target temperature (Ti) in a descending list:
- Instantaneously rescale velocities to Ti.
- Simulate in the NPT ensemble at Ti and 1 bar.
- Monitor the convergence of both potential energy and system density. Continue simulation until the 100-ps running average of both properties changes by less than 0.1% over 500 ps.
- Proceed to next Ti.
- Temperature Steps: Recommended: [500K, 450K, 425K, 415K, 405K, 395K, 385K, 375K, 350K, 300K].
- Analysis: Use the same intersection method as Protocol 1 on data points from the end of each temperature stage.
Visualizations
Workflow: Linear vs Stepwise Cooling for MD
Tg Analysis from V-T Plot Intersection
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Tg Prediction MD |
|---|---|
| Polymer/Small Molecule Topology Files | Defines connectivity, atom types, and bonded parameters for the simulated material (e.g., .itp for GROMACS, .prmtop for AMBER). Critical for accurate chain dynamics. |
| Validated Force Field (e.g., GAFF2, OPLS-AA, CGenFF) | Set of equations and parameters defining interatomic potentials. Choice dictates accuracy of conformational energetics and non-bonded interactions governing Tg. |
| Temperature-Coupling Algorithm (Thermostat) | Maintains correct temperature ensemble. Nosé-Hoover or Bussi-Donadio-Parrinello thermostats are preferred for quenching over simple velocity rescaling. |
| Trajectory Analysis Suite (e.g., MDAnalysis, VMD) | Software to process MD trajectories, calculate volumetric properties, radial distribution functions, and mean squared displacement for relaxation analysis. |
| High-Performance Computing (HPC) Cluster | MD simulations for Tg, especially stepwise protocols, require significant parallel CPU (and sometimes GPU) resources to achieve necessary time scales. |
| Density Analysis Script | Custom or packaged code to compute system volume/density over time, often requiring careful periodic boundary condition handling and averaging. |
Technical Support Center
Troubleshooting Guides
Issue: Glass Transition Temperature (Tg) Drifts with Repeated Simulations
- Problem: Measured Tg is not reproducible between identical cooling runs.
- Diagnosis: This is often caused by insufficient system equilibration at the starting high-temperature state. The system retains a memory of its initial configuration.
- Solution:
- Extend the NPT equilibration at the starting temperature (e.g., 500 K) until energy and density plateau.
- Run multiple independent simulations from different equilibrated starting configurations.
- Use a different random seed for velocity generation.
Issue: Non-Physical Crystallization During Slow Cooling
- Problem: At very slow cooling rates (e.g., 0.1 K/ns), the system crystallizes, which is undesirable for amorphous polymer or glassy system studies.
- Diagnosis: The model system may be too simple or have parameters that favor crystalline ordering.
- Solution:
- Introduce conformational complexity (e.g., atactic polymers, mixtures).
- Apply spatial constraints or use a confining geometry to disrupt long-range order.
- Consider using a cooling rate fast enough to avoid the crystallization timescale.
Issue: High Uncertainty in Extrapolated Tg at Experimental Rates
- Problem: The Vogel-Fulcher-Tammann (VFT) fit to
Tg vs. log(cooling rate)has large confidence intervals when extrapolating to 1 K/min. - Diagnosis: The benchmarked cooling rate range is too narrow or lacks sufficient data points at the slow end.
- Solution:
- Prioritize acquiring more data points in the slower regime (0.1 - 10 K/ns).
- Ensure each cooling rate simulation is repeated 3-5 times for error estimation.
- Cross-validate with an alternative extrapolation method (e.g., Arrhenius fit for fragile systems) to assess robustness.
Frequently Asked Questions (FAQs)
Q1: What is the minimum number of cooling rate decades I should simulate for a reliable extrapolation to experimental rates? A: For credible VFT extrapolation, a range of at least 3-4 orders of magnitude in cooling rate (e.g., from 100 K/ns down to 0.1 K/ns) is recommended. Including a rate near 1 K/ns is critical as it often serves as a useful intermediate anchor point.
Q2: How do I choose the correct property to monitor for Tg determination? A: Specific volume (density) is the most common and robust property. It should be plotted versus temperature and fit with two linear regressions in the high-T (liquid) and low-T (glass) regions. The intersection point is Tg. The coefficient of thermal expansion (α) can also be used, where Tg is marked by a sharp drop in α.
Q3: My simulation box shrinks excessively during cooling. Is this normal? A: Yes, significant volume contraction is expected. To avoid artifacts, always use the NPT ensemble (constant pressure) during the cooling protocol. This allows the box size to adjust naturally and mimics experimental conditions. Ensure your barostat relaxation time is appropriately set (typically 1-5 ps).
Q4: Can I use a non-linear cooling schedule instead of a constant rate? A: While a constant linear cooling rate is the standard for direct benchmarking and comparison to literature, adaptive or staged cooling (fast initially, slower near Tg) can be used to improve efficiency. However, this complicates the direct relationship between rate and Tg and is not recommended for foundational benchmarking studies.
Table 1: Benchmarking Cooling Rates for a Model Polymer (e.g., Polystyrene)
| Cooling Rate (K/ns) | Simulated Tg (± SD) (K) | Computation Time (CPU-hr) | Extrapolated Experimental Tg (K) |
|---|---|---|---|
| 100 | 450 ± 15 | 50 | - |
| 10 | 410 ± 8 | 180 | - |
| 1 | 385 ± 5 | 1,200 | - |
| 0.1 | 375 ± 3 | 12,000 | - |
| VFT Extrapolation to 1 K/min | - | - | 372 ± 5 |
Table 2: Key Metrics for Cooling Rate Selection
| Rate (K/ns) | Primary Use Case | Key Advantage | Major Limitation |
|---|---|---|---|
| 100 - 10 | Rapid screening, qualitative Tg trends, system validation. | Computational efficiency. | Large overestimation of Tg (>50K). |
| 1 | Balance of accuracy and cost; often the main data point for interpolation. | Best practical compromise. | Still requires significant resources. |
| 0.1 | High-accuracy benchmarking, anchoring slow-rate behavior for extrapolation. | Closest to experimental dynamics near Tg. | Prohibitively expensive for large systems. |
Experimental Protocols
Protocol 1: Standard Linear Cooling Simulation for Tg Determination
- System Preparation: Build and fully equilibrate your system (polymer, amorphous drug dispersion, etc.) in the NPT ensemble at a starting temperature well above the expected Tg (e.g., T_start = Tg + 200 K).
- Equilibration: Run a final NPT equilibration for at least 5-10 ns at T_start, monitoring energy and density for stability.
- Cooling Run: Using the NPT ensemble, linearly cool the system from T_start to a final temperature well below Tg (e.g., Tg - 200 K). The cooling rate
Ris defined as(T_start - T_final) / simulation_time. - Data Collection: Save the system volume (and potential energy) at frequent intervals (e.g., every 1-10 ps).
- Analysis: Plot specific volume (or energy) vs. temperature. Perform bilinear fits; the intersection is Tg for that cooling rate
R. - Repeats: Repeat steps 1-5 at least 3 times for each cooling rate
Rusing different initial velocities.
Protocol 2: Multi-Rate Benchmarking for Extrapolation
- Select at least four cooling rates spanning your target range (e.g., 100, 10, 1, 0.1 K/ns).
- For each rate, execute Protocol 1.
- Plot the obtained Tg values against the logarithm of the cooling rate (log₁₀ R).
- Fit the data to the Vogel-Fulcher-Tammann equation:
Tg(R) = T0 + (A / (log₁₀ R - log₁₀ R_inf)), where T0, A, and R_inf are fitting parameters. - Use the fitted equation to extrapolate Tg to the experimental cooling rate (e.g., 1 K/min = 1.67e-11 K/ns).
Visualizations
Title: MD Cooling Protocol for Tg Determination
Title: Tg Dependence on Cooling Rate Log
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in MD Cooling for Tg |
|---|---|
| Force Field (e.g., GAFF2, OPLS-AA, CHARMM) | Defines the potential energy surface; accuracy is critical for realistic chain dynamics and volumetric properties. Must be validated for glassy behavior. |
| Polymer/Amorphous Builder Tool (e.g., PACKMOL, Polymatic) | Generates initial, dense, disordered configurations of complex molecules for simulation, avoiding crystal seeding. |
| Molecular Dynamics Engine (e.g., GROMACS, LAMMPS, NAMD) | Performs the numerical integration of equations of motion. Efficiency and stable NPT implementation are key. |
| Thermostat/Barostat (e.g., Nosé-Hoover, Parrinello-Rahman) | Controls temperature and pressure during equilibration and cooling. A reliable barostat is essential for accurate density. |
| Trajectory Analysis Suite (e.g., VMD/MDTraj, in-house scripts) | Processes simulation output to calculate specific volume, energy, and perform linear regressions for Tg identification. |
| High-Performance Computing (HPC) Cluster | Provides the necessary computational power, especially for slow cooling rates (0.1 K/ns) which require months of real-time simulation. |
Technical Support Center
Troubleshooting Guides
Issue 1: Instability and Energy Explosion
- Symptoms: Simulation crashes with "LINCS warning" or "velocity/force" errors.
- Probable Cause: Timestep too large for the chosen force field and system constraints.
- Solution: Reduce the timestep. For all-atom models with bonds to hydrogen, start with 2 fs. Ensure proper use of constraint algorithms (e.g., LINCS, SHAKE). For coarse-grained models, you may increase to 10-20 fs.
Issue 2: Unphysical System Densities or Lattice Collapse
- Symptoms: Density drifts significantly from experimental values during NPT runs, or the simulation box shrinks uncontrollably.
- Probable Cause: Incorrect pressure coupling settings or barostat choice. Possible mismatch between ensemble and equilibration phase.
- Solution: For NPT, use a semi-isotropic barostat for membrane systems. Ensure sufficient equilibration in NVT before switching to NPT. Adjust the barostat time constant (e.g.,
tau_pin GROMACS) – too short can cause oscillation, too long causes slow equilibration.
Issue 3: Poor Tg Prediction and Rate Dependence Artifacts
- Symptoms: Predicted glass transition temperature (Tg) varies wildly with cooling rate and does not converge.
- Probable Cause: System size too small, leading to excessive finite-size effects. Inadequate sampling across the cooling protocol.
- Solution: Increase system size until Tg predictions stabilize (see System Size Table). Perform multiple independent cooling runs (replicas) and average. Consider using enhanced sampling methods near Tg.
Issue 4: Inconsistent Results Between NVT and NPT Cooling Protocols
- Symptoms: Different density-temperature profiles and derived Tg values when cooling in NVT vs. NPT ensembles.
- Probable Cause: The choice of ensemble physically changes the thermodynamic path of the cooling process.
- Solution: Align ensemble with experimental conditions. For predicting Tg of materials at ambient pressure, an NPT cooling protocol is typically more physically relevant. Always report which ensemble was used.
Frequently Asked Questions (FAQs)
Q1: What is the maximum safe timestep I can use in my cooling simulation? A: The maximum timestep depends on the highest frequency vibration in your system. A common rule is to set it to 1/10th of the period of the fastest motion. For all-atom polymers/drugs with explicit hydrogens, 1-2 fs is standard. Removing hydrogen vibrations via constraints (SHAKE/LINCS) allows 2 fs. For united-atom or coarse-grained systems, 10-30 fs may be possible. Always conduct a short stability test.
Q2: Should I perform my entire cooling simulation in NPT or switch between ensembles? A: A robust protocol involves multiple stages. Typically: 1) Energy minimization, 2) Short NVT equilibration to stabilize temperature, 3) NPT equilibration at the starting high temperature to correct density, 4) Production cooling run in NPT to mimic experimental ambient pressure cooling. Using only NVT during cooling will yield a system at a non-ambient pressure state.
Q3: How do I know if my system is large enough to predict a reliable Tg? A: You must perform a finite-size analysis. Run identical cooling protocols on systems of increasing size (e.g., 2, 4, 8 polymer chains). Plot the predicted Tg vs. 1/(system volume). The point where Tg plateaus indicates a sufficiently large system. For amorphous polymers, systems exceeding 3-5 times the polymer's radius of gyration (Rg) are recommended.
Q4: My cooling rate is 100 K/ns, but experiments are 1 K/min. How can I reconcile this? A: You cannot directly match experimental rates due to computational limits. The key is to perform simulations at multiple, faster cooling rates (e.g., 10, 1, 0.1 K/ns) and extrapolate the predicted Tg to a "zero" cooling rate. This involves a linear regression of Tg vs. log(cooling rate).
Q5: How do pressure coupling parameters affect my NPT cooling simulation?
A: The barostat's time constant (tau_p) is critical. If set too short, it over-corrects box volume, causing large density fluctuations. If too long, the system cannot relax its density efficiently during a rapid cooling quench. For rapid cooling simulations, a tau_p between 10-50 ps is often a practical starting point. The compressibility setting should match the estimated compressibility of your material.
Data Presentation
Table 1: Timestep Guidelines for Cooling Simulations
| Force Field Type | Recommended Timestep (fs) | Key Constraints/Algorithms | Notes for Cooling Runs |
|---|---|---|---|
| All-Atom (w/ H bonds) | 1 - 2 | SHAKE/LINCS for all bonds involving H | Essential for stability during large thermal quenches. |
| United-Atom (no explicit H) | 2 - 4 | LINCS for heavy-atom bonds | More stable at lower T, may allow slight increase. |
| Coarse-Grained (e.g., MARTINI) | 20 - 40 | None for bonds, but may use constraints | Allows faster cooling rates but check density artifacts. |
Table 2: NPT vs. NVT for Tg Prediction
| Parameter | NPT (Constant Number, Pressure, Temperature) | NVT (Constant Number, Volume, Temperature) |
|---|---|---|
| Primary Control | Pressure & Temperature | Volume & Temperature |
| Barostat | Required (e.g., Berendsen, Parrinello-Rahman) | Not Used |
| Density | Fluctuates, determined by P & T | Fixed, may not match experimental pressure |
| Relevance to Experiment | High (mimics ambient pressure cooling) | Low (constant volume cooling is rare) |
| Derived Tg | Typically more accurate | Can be artificially high due to built-in pressure |
Table 3: System Size Impact on Tg Prediction
| System Description | Approx. Atoms/Chains | Predicted Tg Variability (σ) | Recommended for Publication? |
|---|---|---|---|
| Minimal (e.g., 1 short polymer) | < 5,000 atoms / 1-2 chains | Very High (> 15 K) | No – for testing only. |
| Moderate | 10k-50k atoms / 4-10 chains | Moderate (5-10 K) | Yes, with multiple replicas. |
| Well-Converged | > 50k atoms / > 20 chains | Low (< 3 K) | Yes, provides reliable results. |
Experimental Protocols
Protocol 1: Multi-Stage Equilibration Prior to Cooling
- Build & Solvate: Construct initial configuration using PACKMOL or CHARMM-GUI. Ensure proper solvation for drug-polymer systems.
- Energy Minimization: Use steepest descent algorithm for 5,000-10,000 steps until maximum force < 1000 kJ/mol/nm.
- NVT Equilibration (100 ps): Heat system to starting temperature (e.g., 500 K) using a velocity-rescale thermostat (
tau_t= 0.1 ps). Apply position restraints on heavy atoms. Use 2 fs timestep. - NPT Equilibration (200 ps): At starting temperature, equilibrate density using a Berendsen or Parrinello-Rahman barostat (
tau_p= 2-5 ps). Release position restraints. - Production NPT Cooling: Using the equilibrated configuration from step 4, commence the linear cooling protocol to 200 K at the desired rate.
Protocol 2: Finite-Size Analysis for Tg Convergence
- System Preparation: Build 4 systems with increasing size (e.g., 2, 4, 8, and 16 polymer chains). Ensure identical chain length and composition.
- Identical Cooling Protocol: Apply Protocol 1 for equilibration, followed by an NPT cooling run from 500 K to 200 K at a fixed rate (e.g., 1 K/ns) for all systems.
- Density Analysis: For each system, calculate the average density in 5-10 K bins. Fit linear regressions to the high-T (rubbery) and low-T (glassy) regions.
- Tg Determination: Define Tg as the intersection point of the two linear fits.
- Plot & Assess: Plot Tg vs. 1/(Number of Chains) or 1/(Volume). The size where Tg plateaus within error margins is considered sufficient.
Mandatory Visualization
Diagram 1: NPT Cooling Simulation Workflow
Diagram 2: Tg Extrapolation to Zero Cooling Rate
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in MD Cooling Studies |
|---|---|
| GROMACS / LAMMPS / NAMD | Molecular dynamics software engine to perform the simulations. GROMACS is often favored for its speed in sampling. |
| CHARMM36 / GAFF / OPLS-AA | All-atom force fields providing parameters for bonded and non-bonded interactions for polymers, drugs, and biomolecules. |
| MARTINI Force Field | Coarse-grained force field allowing simulation of larger systems and longer timescales, useful for initial screening. |
| VMD / PyMOL / MDAnalysis | Visualization and trajectory analysis toolkits for checking system stability, density profiles, and measuring properties. |
| PACKMOL / CHARMM-GUI | Setup tools for building initial, disordered systems (amorphous cells) and generating necessary topology files. |
| Thermo/Barostat Algorithms | (e.g., Nosé-Hoover, Parrinello-Rahman). Controls temperature and pressure, critical for accurate NPT cooling protocols. |
| LINCS / SHAKE Algorithms | Constraint algorithms that allow longer timesteps by fixing the fastest bond vibrations (e.g., C-H, O-H bonds). |
| Python/Matplotlib | Custom scripting for automated analysis of cooling curves, Tg fitting, and finite-size scaling plots. |
Technical Support Center
Troubleshooting Guides
Issue 1: Job Submission Fails with Cluster Error
- Q: When submitting 100+ cooling rate replicate simulations via my SLURM script, some jobs immediately fail with "PartitionNotAvailable" or "NodeDown" errors. How can I make my submission script more robust?
- A: Implement job submission retry logic and dynamic batch splitting.
- Protocol: Modify your submission script to check the cluster's status before submission. Use
sinfoto parse available partitions and nodes. If the desired resources are unavailable, implement a wait-and-retry loop. For large replicate counts (N>50), split them into smaller batches (e.g., 20 jobs/batch) to avoid overwhelming the scheduler and to allow for partial progress if a cluster issue arises. - Code Snippet Example (Bash):
- Protocol: Modify your submission script to check the cluster's status before submission. Use
Issue 2: Inconsistent Tg Values from Identical Replicates
- Q: My automated analysis script reports a high standard deviation in Tg for replicates that should be identical. What could cause this?
- A: This often stems from non-deterministic simulation initialization or file system I/O conflicts.
- Protocol: Ensure each replicate uses a unique random seed for velocity generation. Verify that temporary file directories (e.g., for MD software like GROMACS or AMBER) are unique per job to prevent overwriting. Implement a post-simulation sanity check in your analysis script to compare the initial configuration energy across replicates.
- Essential Check Table:
Check Command/Tool Expected Outcome Random Seed Uniqueness grep "seed" input*.mdpDifferent value in each input file. Unique Temp Directory echo $MY_TEMP_DIRin job scriptUnique path for each job ID. Initial Potential Energy grep "Potential Energy" replicate_*/energy.logValues within <0.1% of each other.
Issue 3: Automation Pipeline Hangs During Analysis Phase
- Q: The pipeline runs simulations successfully but gets stuck when parsing thousands of output files to calculate density vs. temperature. How can I optimize this?
- A: The issue is likely inefficient I/O operations. Use parallelized analysis and binary file formats.
- Protocol: Replace serial reading of trajectory files with a parallelized library like
MDTrajorMDAnalysis. First, extract only the necessary data (e.g., box volume) into a compact, binary format (HDF5, NumPy.npy) during the simulation run. Then, perform the statistical Tg fitting on this pre-processed data. - Optimized Workflow: Diagram Title: Optimized Analysis Workflow for Tg Calculation
- Protocol: Replace serial reading of trajectory files with a parallelized library like
Frequently Asked Questions (FAQs)
Q1: What is the optimal number of cooling rate replicates for statistically significant Tg prediction in polymer/drug amorphous solid dispersion systems?
- A: Based on recent literature (2023-2024), the required number depends on system size and cooling rate. For typical pharmaceutical polymers (e.g., PVP, HPMC) cooled at 1 K/ns in all-atom simulations:
Always perform a pilot study with 3 replicates to estimate variance before committing to full-scale production runs.System Size (atoms) Suggested Replicates (n) Justification 10,000 - 50,000 8 - 12 Balances statistical power with computational cost for initial screening. 50,000 - 200,000 5 - 8 Larger systems have lower intrinsic variance; fewer replicates are needed. >200,000 3 - 5 Very large systems are computationally expensive; 3-5 replicates are often cited as the minimum.
Q2: How do I choose cooling rates for my MD simulations to reliably extrapolate to experimental conditions?
- A: You must use a minimum of three different simulation cooling rates to perform a meaningful extrapolation. The protocol is:
- Select Rates: Choose rates spanning at least one order of magnitude (e.g., 0.5, 1.0, and 5.0 K/ns).
- Run Replicates: Execute multiple replicates (see Q1) at each rate.
- Calculate Tg(sim): For each replicate, fit the density vs. temperature curve to identify Tg.
- Extrapolate: Plot average Tg(sim) vs. log(cooling rate). Linear extrapolation to the experimental rate (~1 K/min) yields the predicted Tg(exp). Diagram Title: Tg Extrapolation Methodology from MD to Experiment
Q3: What are the critical control parameters to document for each cooling replicate to ensure reproducibility?
- A: Maintain a metadata file (e.g., JSON, YAML) for each replicate with the following:
- Simulation Engine & Exact Version
- Force Field and all modification parameters.
- Initialization Seeds: Random seed for velocities, random number generator seed.
- Cooling Protocol: Starting temperature, final temperature, cooling step size, thermostat (type, coupling constant).
- System Details: Number of atoms, box dimensions, density at starting T.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Cooling Rate Studies for Tg Prediction |
|---|---|
| High-Performance Computing (HPC) Cluster | Provides the parallel processing power required to run tens to hundreds of long-timescale cooling simulations concurrently. |
| Job Scheduler (SLURM/PBS) | Manages resource allocation and job queues for efficient, automated execution of replicate simulations. |
| MD Software (GROMACS, AMBER, LAMMPS) | The core engine for performing the molecular dynamics simulations with precise temperature control. |
| Automation Framework (Python/Bash) | Scripts to automate job submission, file management, error checking, and data collection across all replicates. |
| Parallel Analysis Library (MDTraj, MDAnalysis) | Enables rapid post-processing of large volumes of trajectory data from multiple replicates to extract density/volume vs. temperature. |
| Statistical Software (Python/pandas, R) | Used to perform curve fitting (for Tg per run), statistical aggregation, and linear extrapolation of Tg vs. log(cooling rate). |
| Version Control (Git) | Essential for tracking changes to simulation parameters, scripts, and analysis code to ensure full reproducibility of the study. |
Solving Common Pitfalls: Optimizing Cooling Protocols for Robust Results
Troubleshooting Guide: Identifying Rate-Limited Tg Artifacts
Q1: What are the primary symptoms of a rate-limited, overestimated glass transition temperature (Tg) in molecular dynamics (MD) simulations?
A1: The key symptom is a Tg value that is anomalously high and shows a strong, linear dependence on the applied cooling rate (dT/dt). This is contrary to the physical expectation where Tg should plateau at sufficiently slow cooling rates. Specific indicators include:
- A plot of Tg vs. log(|dT/dt|) is linear with a positive slope, showing no convergence.
- Significant differences in computed Tg (e.g., >20 K) when using different cooling rates like 10 K/ns vs. 100 K/ns.
- Poor agreement with experimental Tg values, even after attempts to map simulation timescales.
Q2: What molecular-level events indicate insufficient equilibration during cooling?
A2: The system fails to sample equilibrium configurational states. Indicators include:
- High Potential Energy: The system gets trapped in a high-energy, non-equilibrium glassy state.
- Low Density: The final glassy structure is less dense than the equilibrium glass.
- Persistent Anisotropy: Residual stress or orientation from the initial liquid state is "frozen in."
- Abrupt Property Changes: Volume or energy vs. temperature shows a sharp kink instead of a smooth transition.
Q3: What is the fundamental protocol to test if my Tg is rate-limited?
A3: Perform a cooling rate study. This is the definitive diagnostic.
Experimental Protocol: Cooling Rate Study
- System Preparation: Start from a well-equilibrated liquid state well above the expected Tg (e.g., Tg + 100 K).
- Cooling Simulations: Run multiple independent NPT (constant Number of particles, Pressure, Temperature) simulations cooling from the high temperature to a low temperature (e.g., Tg - 100 K).
- Vary the Rate: Use at least 4-5 logarithmically spaced cooling rates (e.g., 100 K/ns, 50 K/ns, 10 K/ns, 1 K/ns, 0.5 K/ns). The slowest rate feasible within computational limits is critical.
- Property Tracking: Record potential energy and system volume at regular intervals during cooling.
- Tg Determination: Fit the high-T (liquid) and low-T (glassy) regions of the property vs. T curve to separate linear regressions. Tg is defined as the intersection point.
- Analysis: Plot the calculated Tg against the logarithm of the cooling rate. A plateau indicates a converged Tg; a linear increase indicates a rate-limited artifact.
Table 1: Typical Cooling Rates and Their Impact on Tg for a Small Molecule API
| Cooling Rate (K/ns) | Calculated Tg (K) | Indicator |
|---|---|---|
| 100 | 315 | Severe overestimation, clear artifact |
| 50 | 305 | Overestimation |
| 10 | 290 | Common but often unreliable rate |
| 1 | 282 | Approaching convergence |
| 0.2 | 280 | Likely converged value |
Q4: My cooling rate study shows a linear trend. How do I correct for this artifact?
A4: You must employ strategies to access slower effective cooling rates.
Experimental Protocol: Annealing & Reheating for Tg Convergence
- Slow Cooling: Generate a glassy structure using the slowest cooling rate computationally possible.
- Annealing: Hold the system at a temperature just below the apparent Tg (from the fast run) for an extended time (e.g., 50-100 ns). This allows for local relaxation and densification.
- Reheating Protocol (Key Step): Reheat the annealed glass at a very slow rate (e.g., 0.1-1 K/ns). The Tg measured on this reheating curve is often more reliable because the annealed glass is closer to equilibrium.
- Validation: Compare the Tg from reheating with the trend from your cooling rate study. A value that aligns with the extrapolation to slower rates validates the correction.
FAQ: Practical Considerations for Tg Prediction
Q5: What is the minimum number of cooling rates needed for a reliable study?
A5: A minimum of four distinct rates is required to establish a trend. For publication-quality work, five or more rates spanning at least an order of magnitude in rate are recommended to convincingly demonstrate convergence or quantify the rate dependence.
Q6: Are there computational shortcuts to estimate the equilibrium Tg?
A6: While no perfect shortcut exists, one established method is Fictive Temperature (Tf) analysis.
- Concept: The Fictive Temperature is the temperature at which the glass's configurational properties (like volume) would be in equilibrium. It is equivalent to Tg for a glass formed at equilibrium.
- Protocol: During reheating of a glass, plot the system's property (e.g., enthalpy). The point where the glassy and liquid equilibrium lines intersect during reheating defines Tf. For a well-annealed sample, Tf ≈ Tg.
Q7: How do I choose the right property to track for Tg determination?
A7: Volume and potential energy are most common. Use both for robustness.
- Volume (V): Often gives a clearer signal in NPT simulations. More directly comparable to experimental dilatometry.
- Potential Energy (U): Sensitive to both intermolecular and intramolecular degrees of freedom. Can be noisier.
- Best Practice: Calculate Tg from both V(T) and U(T). The values should agree within a reasonable margin (e.g., ±5 K). A large discrepancy may indicate other issues.
Diagram Title: Workflow for Diagnosing and Correcting Rate-Limited Tg
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Components for Robust Tg Simulation Studies
| Item | Function & Rationale |
|---|---|
| Validated Force Field (FF) | The foundation. Must accurately reproduce intermolecular interactions (van der Waals, Coulombic) and torsional potentials. Use pharmaceutical FF (e.g., GAFF2, OPLS4) with validated partial charges. |
| Long-Timescale MD Engine | Software capable of stable, GPU-accelerated NPT dynamics for µs-scale simulations (e.g., GROMACS, OpenMM, NAMD, DESMOND). |
| Robust Thermostat/Barostat | Controls temperature and pressure without artifacts. Nose-Hoover thermostat + Parrinello-Rahman barostat or MTK barostat are standards for NPT. |
| Automated Analysis Scripts | Custom Python/MATLAB scripts for batch processing cooling runs, performing linear fits, intersection detection (Tg calculation), and plotting Tg vs. log(rate). |
| Enhanced Sampling Plugins (Optional) | Tools like PLUMED can be used to implement metadynamics or parallel tempering to improve sampling near Tg for small systems. |
| High-Performance Computing (HPC) Allocation | Essential resource. Running multiple slow cooling replicates requires thousands of GPU/core hours. |
Diagram Title: Logical Relationship: Cooling Rate to Tg Outcome
Troubleshooting Guides
Q: What are the primary causes of high variability in Tg predictions from cooling rate simulations? A: High variability typically stems from three main areas:
- Insufficient Sampling: The simulation time or number of independent replicas is too low to adequately sample the phase space.
- Inconsistent Equilibration: Incomplete or uneven equilibration before the cooling cycle begins, leading to different starting points.
- Parameter Sensitivity: Small variations in force field parameters or cooling protocol details (e.g., step size, thermostat coupling) amplify over the simulation.
Diagnostic Steps:
- Run 5-10 independent simulation replicas from different initial velocities.
- Plot the specific volume vs. temperature for each replica on the same graph.
- Calculate the standard deviation of the predicted Tg (from the intersection of linear fits) across replicas.
- If the standard deviation exceeds 5-10 K, proceed to the protocol optimization in Guide 2.
Guide 2: Protocol for Statistically Robust Tg Determination
Q: What is a step-by-step method to reduce variability and ensure significance? A: Follow this standardized protocol to minimize inter-replica differences.
Experimental Protocol:
- System Preparation: Build and minimize your amorphous polymer/drug system.
- NPT Equilibration: Equilibrate at 50-100 K above the expected Tg for a minimum of 10 ns. Monitor density/potential energy for stability.
- Replica Generation: Generate N independent replicas (N≥10 for statistical rigor) by assigning new random velocity seeds from the Maxwell-Boltzmann distribution at the equilibration temperature.
- Cooling Cycle: Apply a linear cooling ramp (e.g., from 500 K to 200 K) at a defined rate (e.g., 1 K/ns) identically to all replicas. Use a consistent thermostat (e.g., Nosé-Hoover) and barostat.
- Data Collection: Record specific volume (or enthalpy) every 1-2 K.
- Analysis:
- For each replica, perform a piecewise linear regression on the specific volume vs. temperature data.
- Identify Tg (replica) as the intersection point of the high-T and low-T linear fits.
- Calculate the mean Tg, standard deviation (SD), and standard error of the mean (SEM = SD/√N) across all replicas.
- Report Tg as Mean Tg ± SEM and state N.
Frequently Asked Questions (FAQs)
Q: How many replicate simulations (N) are statistically sufficient for Tg prediction? A: There is no universal number, but a power analysis can determine N. As a rule of thumb, start with N=10. Use the initial results to calculate the required N to achieve a desired confidence interval (e.g., ±3 K) using the formula related to the t-distribution: n ≈ (tσ / ME)²*, where t is the t-statistic, σ is the estimated SD, and ME is the desired margin of error.
Q: How do I differentiate between true thermodynamic glass transition variability and artifacts from poor simulation settings? A: Conduct this control experiment: Run 5 replicas at an extremely slow cooling rate (e.g., 0.1 K/ns). If variability remains high, the issue is likely insufficient equilibration or force field inaccuracy. If variability decreases significantly, your original cooling rate was too fast, causing non-equilibrium artifacts.
Q: Which statistical test is most appropriate for comparing Tg values from two different cooling rates or formulations? A: Use an unpaired, two-sample t-test (assuming normally distributed Tg values from replicates). First, perform an F-test to check for equal variances between the two groups. If variances are not equal, use Welch's corrected t-test. Report the p-value alongside the mean difference and confidence intervals.
Data Presentation
Table 1: Impact of Replica Count (N) on Tg Prediction Confidence for a Model Polymer (Hypothetical Data)
| Cooling Rate (K/ns) | N (Number of Replicas) | Mean Tg (K) | Standard Deviation (SD) | Standard Error of Mean (SEM) | 95% Confidence Interval (K) |
|---|---|---|---|---|---|
| 1.0 | 5 | 325.1 | 8.7 | 3.9 | 321.2 - 329.0 |
| 1.0 | 10 | 323.8 | 7.2 | 2.3 | 321.5 - 326.1 |
| 1.0 | 20 | 324.3 | 6.5 | 1.5 | 322.8 - 325.8 |
| 0.1 | 10 | 338.5 | 2.1 | 0.7 | 337.8 - 339.2 |
Table 2: Statistical Comparison of Tg at Two Different Cooling Rates (N=10 each)
| Parameter | Cooling Rate: 1.0 K/ns | Cooling Rate: 0.1 K/ns |
|---|---|---|
| Mean Tg (K) | 323.8 | 338.5 |
| Standard Deviation (K) | 7.2 | 2.1 |
| Pooled Variance | 28.6 | - |
| t-statistic (unpaired) | 6.12 | - |
| p-value | < 0.0001 | - |
| Conclusion | The difference in Tg is statistically significant. |
Experimental Protocols
Detailed Protocol: Multi-Replica Cooling Simulation for Tg
- Initial System Builder: Use PACKMOL or CHARMM-GUI to build an amorphous cell with ~100 polymer chains or drug molecules.
- Energy Minimization: Apply 5,000 steps of steepest descent followed by 5,000 steps of conjugate gradient algorithm.
- Equilibration in NVT: Heat system to 100 K above expected Tg using Langevin thermostat for 1 ns.
- Equilibration in NPT: Run at target starting temperature (T_start) for 10 ns using a Nosé-Hoover thermostat and Parrinello-Rahman barostat. CRITICAL: Ensure density fluctuation is less than 1% over the final 2 ns.
- Replica Seed Generation: From the final equilibrated structure, generate
Nconfiguration files, each with randomized velocities using a different random seed. - Cooling Simulation: For each replica, run an NPT simulation while linearly decreasing temperature from Tstart to Tend over the defined simulation time. Pressure remains constant at 1 atm. Log thermodynamic data every 1000 steps.
- Post-Processing: Use scripts (Python, VMD) to calculate specific volume (box volume / mass) per frame. Align data from all replicas for analysis.
Visualizations
Title: MD Replica Workflow for Robust Tg
Title: Statistical Significance Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Software for MD Cooling Studies
| Item Name | Category | Function/Brief Explanation |
|---|---|---|
| GROMACS | Software | High-performance MD engine; optimal for running many replica simulations in parallel due to its efficiency. |
| AMBER/CHARMM | Software/Force Field | Provides highly validated force field parameter sets for organic molecules and polymers crucial for accurate Tg prediction. |
| Python (MDAnalysis, SciPy) | Analysis Scripting | Essential for automating trajectory analysis, piecewise linear fitting of Vol vs. Temp data, and statistical calculations across replicas. |
| High-Performance Computing (HPC) Cluster | Hardware | Necessary computational resource to run multiple long-timescale replica simulations concurrently. |
| Nosé-Hoover Thermostat | Simulation Parameter | Provides correct canonical (NVT) or isothermal-isobaric (NPT) ensemble sampling, crucial for temperature control during cooling. |
| Parrinello-Rahman Barostat | Simulation Parameter | Allows cell fluctuations during NPT equilibration and cooling, critical for accurate density and specific volume calculation. |
| Polymer/Drug Specific Force Field (e.g., GAFF, CGenFF) | Research Reagent | Pre-parameterized atomistic force fields for simulating small molecule pharmaceuticals or polymer components. |
| Visual Molecular Dynamics (VMD) | Visualization Software | Used to inspect system equilibration, check for crystallization artifacts, and render simulation snapshots for publication. |
Troubleshooting Guides & FAQs
Q1: My system's potential energy plateaus during cooling, but the Tg value I predict varies significantly between cooling rates. Is the system truly equilibrated at each temperature step? A: A potential energy plateau is necessary but not sufficient for verifying equilibration in the glassy state. For reliable Tg prediction, you must check for the cessation of structural relaxation. Use the following protocol:
- At each temperature step after the energy plateau is reached, calculate the mean squared displacement (MSD) of the alpha-carbons (or backbone atoms for polymers) over a time window equal to 5x the equilibration time at that step.
- The system is equilibrated when the MSD vs. time plot on a log-log scale shows a slope << 1 (approaching 0), indicating sub-diffusive, arrested motion.
- If the slope remains near 0.6, structural relaxation is ongoing; extend the equilibration time.
Q2: How long should I equilibrate at each temperature during a cooling run for Tg prediction? A: There is no universal time; it depends on your material and proximity to Tg. Use a convergence criterion, not a fixed time. Implement this protocol:
- Perform block averaging of the system's density (or enthalpy) over consecutive time blocks during the equilibration phase at a given temperature.
- Start with a block size of 10 ps and increase gradually.
- Equilibration is achieved when the standard error between block averages is less than 1% of the mean value. The table below summarizes recommended checks:
| Check | Metric | Target for Equilibration | Typical Time Scale (near Tg) |
|---|---|---|---|
| Energy | Total Potential Energy | Plateau (slope ≈ 0) | 1-10 ns |
| Structure | Radial Distribution Function (g(r)) | Convergence of first peak height/area | 2-20 ns |
| Dynamics | Mean Squared Displacement (MSD) | Log-log slope << 1 (arrested) | 10-100 ns |
| Convergence | Block Average of Density | Standard error < 1% of mean | 50+ ns |
Q3: I see jumps in the volume vs. temperature plot at slow cooling rates. Is this physical or a sign of poor equilibration? A: This is likely a sign of partial equilibration. At very slow cooling rates, the system has more time to find lower energy configurations, which can cause discrete "drops" in volume. This is physical but problematic for defining a single Tg. To mitigate:
- Protocol: Perform 3-5 independent cooling runs from different equilibrated liquid configurations.
- Calculate Tg for each run (via intersection of linear fits).
- Report the mean and standard deviation of Tg. A large standard deviation (>5 K) indicates your cooling rate is too slow for reproducible analysis with your chosen equilibration times.
Q4: What are the best order parameters to confirm a glass has formed, not a crystal? A: Use a combination of structural and dynamical metrics.
- Radial Distribution Function (RDF): Compare the final cooled state to the initial liquid and a known crystal structure. A glass will retain the broad, split-second peak of the liquid, while a crystal will show sharp, new peaks.
- Bond-Orientational Order Parameters (Steinhardt-Nelson Q₆): Calculate the global Q₆ parameter. It will remain low (~0.1-0.3) for a glass but jump to a high value (>0.5) for a crystal.
- Visual Inspection: Use VMD or PyMOL to visually inspect for long-range periodic order.
The Scientist's Toolkit: Research Reagent Solutions
| Item / Software | Function in Tg/MD Research |
|---|---|
| GROMACS / LAMMPS / AMBER | Molecular dynamics engines to perform the cooling simulations. GROMACS is often preferred for its speed in NVT/NPT ensembles. |
| MDAnalysis / VMD | Analysis toolkits for calculating MSD, RDF, density, and energy trends from trajectory files. |
| Python (NumPy, SciPy, Matplotlib) | Essential for scripting custom analysis, block averaging, curve fitting (for Tg intersection), and plotting results. |
| PyPRISM | Calculates structure factors and sophisticated order parameters beyond simple RDF. |
| Force Field (e.g., OPLS-AA, CHARMM, GAFF) | The interatomic potential model. Selection is critical; it must be validated for glass-forming systems (density, Tg trend). |
| Glass-Forming System (e.g., Sorbitol, Amorphous Drug Compound) | The molecule of interest. Often a small organic with a known experimental Tg for method validation. |
| High-Performance Computing (HPC) Cluster | Necessary for long timescale (µs+) simulations at slow cooling rates (e.g., 0.01 K/ns). |
Experimental Workflow & Pathway Diagrams
Title: Equilibration Check Workflow for MD Cooling
Title: Protocol for Tg Prediction from MD Cooling Rates
Technical Support Center: Troubleshooting & FAQs
FAQ: General System Size Concerns
Q1: How do I know if my simulation box is too small for a reliable Tg prediction? A: A primary symptom is a significant dependence of the calculated Tg on the system size (number of molecules, N) or box length (L). If doubling your system size changes Tg by more than 2-5 Kelvin, your original box is likely too small. Other signs include:
- Artificially high density or pressure due to periodic images interacting.
- Suppression of long-wavelength fluctuations critical for the glass transition.
- Abnormal dynamic heterogeneity profiles.
Q2: What specific finite-size effects plague cooling rate studies in MD simulations? A: The main coupling is between system size (L) and the effective cooling rate (q). A smaller box can artificially accelerate or alter the dynamics, making the system appear to vitrify at a different temperature. This confounds the extrapolation to experimental cooling rates. You may observe:
- Size-Dependent Tg(q): The Tg you measure at a given cooling rate shifts with box size.
- Altered Relaxation Times: The finite box truncates the spectrum of relaxation modes, affecting the calculation of the Vogel-Fulcher-Tammann parameters.
- Anisotropic Dynamics: Dynamics may differ along box vectors if the box is not large enough to be isotropic.
Q3: What is a practical protocol to test for finite-size effects in my Tg study? A: Follow this systematic protocol:
- Define Property: Choose your primary indicator for Tg (e.g., specific volume V(T), enthalpy H(T)).
- Base Simulation: Run your standard cooling protocol (e.g., 1 K/ns) for your initial/typical system size (N0, L0).
- Size Variation: Repeat the exact same cooling protocol for systems with 2x, 4x, and 8x the number of molecules (maintaining density). For polymeric systems, also consider varying chain length independently of box size.
- Analysis: Fit the high-T and low-T linear regimes of V(T) or H(T) for each run. Determine Tg for each system size (N).
- Extrapolation: Plot Tg(N) vs. 1/N (or Tg(L) vs. 1/L). Extrapolate to 1/N → 0 (infinite system size). The point of convergence indicates the size-independent Tg.
Experimental Protocol: Finite-Size Scaling for Tg
Objective: To determine the system size (N) required for a Tg estimate invariant within a target precision (e.g., ±1 K) for a specific polymer/drug formulation.
Methodology:
- System Preparation: Build initial configuration of your molecule(s) using Packmol or similar. Use a minimum of 3 distinct system sizes (e.g., 20, 40, 80 chains for polymers; 500, 2000, 8000 molecules for simple glass formers).
- Equilibration: For each size (N), perform NPT equilibration at T > Tg (experimental + 100 K) for >20 ns. Ensure pressure ~1 atm and monitor density convergence.
- Cooling Runs: Apply identical, linear cooling ramps from the equilibrated high T to a low T (well below expected Tg) for all systems. Use the same thermostat/barostat settings. Example: Cool from 500 K to 200 K at a rate of 0.5 K/ns.
- Data Collection: Output specific volume (V) and enthalpy (H) at frequent intervals (every 1-5 K drop).
- Tg Determination: For each size N, perform a two-regime linear fit to V(T) or H(T). Tg is the intersection point of the high-T (liquid) and low-T (glass) lines.
- Finite-Size Analysis: Plot the obtained Tg(N) against 1/N. Perform a linear or quadratic fit. The y-intercept (1/N → 0) is your size-corrected Tg(∞).
Data Presentation: Finite-Size Scaling Results for a Model Polymer (Hypothetical Data)
Table 1: Tg Dependence on System Size for Poly(styrene) at 1 K/ns Cooling Rate
| Number of Chains (N) | Total Atoms | Box Length (Å) | Calculated Tg (K) | Density at 300K (g/cm³) |
|---|---|---|---|---|
| 10 | ~16,000 | 45.2 | 362.1 ± 3.5 | 1.032 |
| 20 | ~32,000 | 56.9 | 370.3 ± 2.1 | 1.045 |
| 40 | ~64,000 | 71.7 | 373.8 ± 1.8 | 1.049 |
| 80 | ~128,000 | 90.3 | 375.5 ± 1.2 | 1.051 |
| Extrapolated (N→∞) | ∞ | ∞ | 376.8 ± 0.7 | 1.052 |
Table 2: Key Artifacts vs. System Size
| Artifact/Symptom | Severe (N=10) | Moderate (N=20) | Minimal (N=80) |
|---|---|---|---|
| Tg Shift vs. N→∞ | +14.7 K | +6.5 K | +1.3 K |
| Density Error % | -1.9% | -0.7% | -0.1% |
| Anisotropy (Pxx/Pyy) | 1.15 | 1.05 | 1.01 |
| α-Relaxation Time Error* | ~35% | ~15% | <5% |
*Estimated from box-size effect on longest wavelength modes.
Mandatory Visualizations
Title: Finite-Size Testing Protocol for Tg Studies
Title: Coupling of Finite-Size and Finite-Cooling-Rate Effects
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Finite-Size Analysis
| Item/Software | Function in Finite-Size Analysis | Key Consideration |
|---|---|---|
| MD Engine (GROMACS, LAMMPS, OpenMM) | Performs the core cooling simulations. Enables parallel computation for larger systems. | Optimize neighbor searching and PME settings for large boxes. Monitor performance scaling with N. |
| System Builder (Packmol, Moltemplate) | Creates initial configurations for multiple system sizes at target density. | Ensure consistency in packing quality and randomness across different sizes. |
| Python/NumPy/SciPy | For automated analysis of trajectories, linear fitting for Tg, and finite-size scaling plots. | Script the entire analysis pipeline for reproducibility across all system sizes. |
| Visualization (VMD, OVITO) | To visually inspect configurations for crystallization artifacts or anisotropic structures. | Critical for diagnosing packing failures in small boxes. |
| CLAYFF, GAFF, OPLS-AA | Force fields parameterizing interactions. Finite-size effects can be force-field dependent. | Validate force field performance (e.g., density) at all system sizes before long cooling runs. |
| High-Performance Computing (HPC) Cluster | Provides the necessary computational resources to run multiple, large-scale cooling simulations. | Plan resource allocation: Larger N reduces finite-size error but increases cost per simulation. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My simulation fails to reach equilibrium during the adaptive cooling phase, and the system energy diverges. What could be the cause? A: This is typically caused by an excessively high initial cooling rate or an improper coupling algorithm. Ensure your initial cooling rate (e.g., 1 K/ns) does not exceed the system's relaxation capacity. Use the Berendsen thermostat for initial equilibration before switching to Nosé–Hoover for the adaptive phase. Verify that your timestep (1-2 fs) is appropriate for your force field and constraints.
Q2: How do I diagnose poor glass transition (Tg) prediction from my hybrid simulation output? A: Poor Tg prediction often stems from insufficient sampling or an incorrect cooling schedule. First, plot the specific volume vs. temperature. A clear inflection point should be visible. If the curve is smooth, increase the sampling time at each temperature window by 20-50%. Use the following diagnostic table:
| Symptom | Likely Cause | Recommended Action |
|---|---|---|
| Smooth V vs. T curve | Inadequate sampling | Increase simulation time per temperature; replicate runs. |
| High variance in Tg (>10 K) | Poor statistical averaging | Perform ≥ 5 independent runs; use block averaging. |
| Tg deviates from experimental >15% | Force field mismatch | Validate force field with known density at a reference T. |
| Discontinuous volume drop | Cooling rate too high | Implement adaptive cooling; reduce max dT/dt. |
Q3: The hybrid MD/Monte Carlo (MC) simulation is stalling—acceptance rate for MC moves is nearly zero. How do I fix this? A: A low acceptance rate indicates the MC move steps are too large for the current dense, low-temperature phase. Implement an adaptive move step size. Reduce the maximum displacement or rotation angle by 50% when the acceptance rate falls below 15%. Revert to original parameters if the temperature increases in an adaptive protocol.
Q4: When using adaptive cooling, how do I program the feedback loop to adjust the rate effectively? A: The feedback should monitor the rate of energy change (dE/dt). Use this protocol:
- Define a target relaxation metric, e.g.,
τ_target = 10 ps. - At each feedback interval (e.g., every 5 ps), calculate the actual relaxation time
τ_actualfrom an exponential fit of the energy autocorrelation. - Adjust cooling rate:
new_rate = current_rate * (τ_target / τ_actual). - Set bounds: e.g.,
0.01 K/ns ≤ new_rate ≤ 5 K/ns.
Detailed Experimental Protocol: Optimizing Cooling Rates for Tg Prediction
Title: Protocol for Hybrid MD/MC Simulation with Adaptive Cooling for Tg Determination.
Objective: To accurately predict the glass transition temperature (Tg) of an amorphous polymer/drug dispersion using an adaptive cooling schedule within a hybrid simulation framework.
Materials & System Setup:
- Software: GROMACS/NAMD with PLUMED for MD; in-house MC module.
- Initial Configuration: 100 polymer chains (50 monomers each) + 50 drug molecules, packed via PACKMOL.
- Force Field: CHARMM36/GAFF with RESP charges.
- Solvent: Implicit solvent (GBSA) or explicit (TIP3P) with periodic boundaries.
Procedure:
- High-T Equilibration: Energy minimize. Run NPT at 500 K (100 K above estimated Tg) for 50 ns. Confirm equilibrium via stable density/Rg.
- Hybrid Simulation Initiation:
- Set initial cooling rate to 1.0 K/ns.
- Define a cooling cycle: For every 1 ns of MD (NPT, Nosé–Hoover thermostat, Parrinello-Rahman barostat), attempt 1000 MC moves (translation, rotation, conformational swap) per molecule.
- Adaptive Cooling Implementation:
- Every 10 K decrement, pause simulation.
- Perform a short 20 ps NVE run to compute the enthalpy relaxation function.
- If relaxation time
τ > 15 ps, reduce cooling rate by factor of 0.8 for the next window. - If
τ < 5 ps, increase cooling rate by factor of 1.2.
- Data Collection: Output specific volume, enthalpy, and radial distribution functions every 1 ps. Record accepted MC moves.
- Cooling & Analysis: Cool system to 200 K. Perform heating run at a constant slow rate (0.5 K/ns) to check for hysteresis. Fit specific volume data from cooling cycle with two linear regressions; Tg is the intersection point.
Visualization: Workflow and Logic
Title: Adaptive Cooling Hybrid Simulation Workflow
Title: Adaptive Cooling Feedback Loop Logic
The Scientist's Toolkit: Research Reagent Solutions
| Item Name | Function in Experiment | Key Consideration |
|---|---|---|
| CHARMM36 Force Field | Provides parameters for bonding/non-bonded interactions for polymers/biomolecules. | Use C36m version for better disordered phase accuracy. |
| GAFF with RESP Charges | General Amber Force Field for small molecule drug parameterization. | Ensure charges fitted at relevant pH using quantum chemistry. |
| TIP3P Water Model | Explicit solvent model for solvated systems. | May overestimate diffusion; consider TIP4P/2005 for denser glass. |
| PACKMOL Software | Generates initial configuration of mixed molecules in a simulation box. | Requires careful input of molar ratios and target density. |
| PLUMED Plugin | Enables enhanced sampling and collective variable analysis. | Essential for implementing custom feedback on relaxation metrics. |
| Nosé–Hoover Thermostat | Produces correct canonical (NVT) ensemble dynamics. | Use chains (NHC) to avoid non-ergodic behavior in long runs. |
| Parrinello-Rahman Barostat | Allows isotropic/anisotropic box fluctuations in NPT ensemble. | Coupling time constant should be ~4x the thermostat constant. |
| Hybrid MC Move Set | Enables large conformational sampling alongside MD. | Includes translation, rotation, and dimer/swap moves for efficiency. |
Benchmarking Against Reality: Validating MD-Predicted Tg with Experimental Data
Technical Support & Troubleshooting Center
FAQ 1: Why does my simulated Tg from MD cooling runs differ significantly from my experimental DSC measurement?
- Answer: Discrepancies are common and often stem from cooling rate differences. MD simulations typically use rates of 1-100 K/ns, while DSC uses 0.083–1.67 K/s (5–100 K/min). This is a ~10^9 to 10^11 fold difference. Use the Vogel–Fulcher–Tammann (VFT) equation or the relationship
log(R_sim) = A - B/(Tg_sim - T0)to extrapolate your MD-derived Tg to experimental rates. Ensure your force field is validated for glass-forming systems; inaccurate van der Waals or torsional potentials can skew results.
FAQ 2: My MD-calculated specific heat capacity (Cp) jump at Tg does not match the DSC thermogram. How can I improve correlation?
- Answer: The Cp calculation method in MD is critical. Ensure you are calculating it correctly via energy fluctuations:
Cp = (⟨H²⟩ - ⟨H⟩²) / (k_B T² N), where H is enthalpy. Common pitfalls include:- Insufficient equilibration: The system must be fully equilibrated above and below Tg for accurate ensemble averages.
- Small system size: Finite size effects can smear the transition. Use >10,000 atoms if possible.
- Short sampling time: The simulation time must be long enough to capture slow relaxation near Tg. Perform block averaging to check convergence.
FAQ 3: How do I properly define the Tg from my MD cooling trajectory to match the DSC midpoint method?
- Answer: DSC typically reports Tg at the midpoint of the Cp step transition. To replicate this in MD:
- Perform a cooling run, saving potential energy or volume at regular temperature intervals.
- Fit the high-T (liquid) and low-T (glassy) data regions with separate linear regressions.
- Identify the intersection point of these two lines. This temperature is your simulated Tg, analogous to the DSC midpoint. Do not use the onset or endpoint without specifying the method.
FAQ 4: I am simulating a polymer or amorphous drug. What protocol ensures the most representative initial configuration for DSC comparison?
- Answer: Follow this validated protocol:
- Build & Solvate: Use PACKMOL or similar to create an amorphous cell with correct experimental density (±5%).
- High-T Equilibration: Run in the NPT ensemble at 50-100 K above the expected Tg for at least 10-100 ns to erase memory of initial configuration.
- Density Validation: Check the final density matches the experimental (or theoretical) value for your force field.
- Cooling Procedure: Begin your production cooling run from this well-equilibrated, high-temperature state.
Table 1: Cooling Rate Effect on Simulated Glass Transition Temperature (Tg)
| Material (Example) | MD Cooling Rate (K/ns) | MD-derived Tg (K) | Extrapolated to DSC rate (0.167 K/s) (K) | Experimental DSC Tg (K) | Reference Force Field |
|---|---|---|---|---|---|
| Amorphous Sucrose | 1 | 315 | 331 | 332 | GLYCAM06/general AMBER |
| Atactic Polystyrene | 10 | 390 | 373 | 375 | OPLS-AA |
| Indomethacin (γ-form) | 0.5 | 315 | 329 | 330-332 | GAFF2 |
Table 2: Key Metrics for MD-DSC Correlation
| Metric | How it's Obtained from MD | Corresponding DSC Readout | Critical Parameters for Match |
|---|---|---|---|
| Glass Transition Temp (Tg) | Intersection of fitted lines for V or U vs T. | Midpoint of Cp step change. | Cooling rate, definition of linear regions. |
| Heat Capacity Jump (ΔCp) | Fluctuation formula using enthalpy (H). | Step height in Cp curve. | Sufficient sampling, accurate H calculation. |
| Thermal Expansion Coeff. (α) | Slope of V vs T plot above and below Tg. | Dilatometry data; inferred from DSC. | NPT ensemble stability, averaging. |
Experimental & Simulation Protocols
Protocol 1: Standard DSC Measurement for Tg Determination
- Sample Preparation: Weigh 5-10 mg of amorphous material into a hermetic aluminum DSC pan. Crimp the lid to ensure an airtight seal.
- Instrument Calibration: Calibrate the DSC cell for temperature and enthalpy using indium and zinc standards.
- Method Programming:
- Equilibrate at 20°C below expected Tg.
- Ramp temperature at standard rate (e.g., 10 K/min) to 30°C above expected Tg under nitrogen purge (50 mL/min).
- Cool back to start at same rate.
- Run a second heating cycle; analyze this cycle to remove thermal history.
- Data Analysis: Plot heat flow vs. temperature. Tg is taken as the midpoint of the step change in heat flow on the second heating scan.
Protocol 2: MD Cooling Simulation for Tg Prediction
- Initial System: Start from a well-equilibrated, high-temperature (e.g., Tg + 100K) NPT simulation.
- Cooling Run Setup: Using NPT ensemble, linearly decrease the temperature from the high-T state to well below the expected Tg over the course of the simulation. (e.g., from 500K to 200K over 500 ns = 0.6 K/ns).
- Data Collection: Save system volume and potential energy at frequent intervals (e.g., every 1-10 ps).
- Analysis:
- Plot average volume (or energy) per atom vs. temperature.
- Perform linear fits to the high-T (rubbery/liquid) and low-T (glassy) data points.
- Calculate Tg as the intersection temperature of the two linear fits.
Visualizations
Title: MD Simulation Workflow for Tg Prediction
Title: MD-DSC Correlation Pathway
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function/Description |
|---|---|
| Hermetic Aluminum DSC Pans & Lids | Seals sample during DSC run to prevent solvent loss and ensure stable baseline. Critical for amorphous materials. |
| Indium & Zinc Calibration Standards | High-purity metals for calibrating DSC temperature and enthalpy scales, ensuring measurement accuracy. |
| High-Performance Force Fields (e.g., GAFF2, OPLS-AA, CHARMM36) | Parameter sets defining atom interactions in MD. Choice is critical for accurate density, energy, and Tg prediction. |
| Amorphous Cell Building Tools (PACKMOL, Molten Salt Builder) | Software to create realistic, random initial configurations of molecules for amorphous system MD simulations. |
| VFT Equation Fitting Script | Custom or published script to extrapolate Tg from MD-rate to experimental-rate using the Vogel–Fulcher–Tammann relationship. |
| Nitrogen Gas (High Purity) | Inert purge gas for DSC to prevent sample oxidation and maintain a clean furnace during heating/cooling cycles. |
Troubleshooting Guides & FAQs
Q1: My MD-simulated Tg is consistently lower than the experimental DSC value for indomethacin. What are the primary causes? A: This is often due to an excessively fast cooling rate in the simulation. Molecular dynamics simulations typically cool systems at rates of ~1-100 K/ns, which are many orders of magnitude faster than experimental DSC rates (~0.1-10 K/min). This prevents adequate structural relaxation, leading to an under-predicted Tg. Solution: Implement a stepwise cooling protocol with extended equilibration periods at each temperature or employ a replica-exchange method to enhance sampling.
Q2: During the cooling simulation of itraconazole, the density-temperature plot shows high scatter, making Tg fitting difficult. How can I improve the data quality? A: High scatter indicates insufficient equilibration and/or poor statistical sampling. Solution: 1) Increase the simulation time at each temperature step (aim for >5-10 ns of production MD per point after equilibration). 2) Use a larger simulation box to reduce fluctuations. 3) Run multiple independent replicas and average the density values. 4) Apply a suitable moving average or linear regression fit to the data before identifying the Tg intersection point.
Q3: The Tg prediction for my amorphous drug system is highly sensitive to the force field choice. Which force fields are most reliable for pharmaceuticals like indomethacin? A: Force field accuracy is critical. The OPLS-AA/GAFF families are widely used and show good performance for organic molecules. For specific systems:
- Indomethacin: The GAFF2 force field with AM1-BCC charges, coupled with the TIP3P water model for solvated systems, has yielded predictions within 5-10 K of experiment when using optimized cooling rates.
- Itraconazole: Due to its complexity, OPLS-AA with manually curated torsional parameters for flexible dihedrals is recommended. Always validate force field performance by comparing simulated densities and radial distribution functions with available experimental or ab initio data.
Q4: What is the most robust method to extract Tg from the simulated data (V/T or E/T plots)? A: While both specific volume (V) and potential energy (E) versus temperature (T) are used, specific volume is generally more robust for Tg determination in MD. Energy plots can be noisier. The standard protocol is:
- Plot specific volume (or enthalpy) against temperature.
- Perform a piecewise linear regression on the high-T (liquid) and low-T (glass) data regions.
- Define Tg as the intersection point of the two fitted lines. Statistical uncertainty can be assessed by bootstrapping the data points.
Q5: How long should I equilibrate the system at each temperature step during cooling? A: Equilibration time must be validated. A common and effective protocol is:
- At each new temperature, run NPT equilibration until the density and energy plateau.
- Monitor the running average of the density; equilibration is typically achieved when the drift is less than 0.1% over a 1 ns window.
- As a rule of thumb, for small-molecule glasses, allocate 2-5 ns for equilibration in the supercooled liquid region and 5-10 ns closer to and below Tg.
Table 1: Experimental vs. Simulated Tg Values for Model Systems
| Compound | Experimental Tg (K) [DSC] | Simulated Tg (K) [MD] | Cooling Rate (MD) | Force Field | Error (K) |
|---|---|---|---|---|---|
| Indomethacin | 315 - 318 | 305 - 310 | 0.5 K/ns | GAFF2 | -8 to -5 |
| Indomethacin | 315 - 318 | ~300 | 10 K/ns | GAFF1 | -15 to -18 |
| Itraconazole | 330 | 322 - 326 | 0.25 K/ns | OPLS-AA (mod.) | -5 to -8 |
Table 2: Impact of MD Cooling Rate on Predicted Tg (Indomethacin)
| MD Cooling Rate (K/ns) | Predicted Tg (K) | Density Slope (Liquid) [g/cm³K] | Equilibration Time per Step (ns) |
|---|---|---|---|
| 10 | 298 | -2.1e-4 | 1 |
| 1 | 307 | -2.4e-4 | 2 |
| 0.1 | 312 | -2.9e-4 | 20 |
Experimental Protocols
Protocol 1: Standard Stepwise Cooling MD for Tg Prediction
- System Preparation: Build an amorphous cell of the drug molecule (e.g., 100-500 molecules) using PACKMOL or similar software.
- Energy Minimization: Minimize the structure using steepest descent/conjugate gradient algorithm (5000 steps).
- Initial Equilibration: Equilibrate in the NPT ensemble at 50 K above the expected Tg (e.g., 370 K for indomethacin) for 10 ns. Use a Berendsen or Parrinello-Rahman barostat (1 atm) and a Nosé-Hoover thermostat.
- Stepwise Cooling: Reduce temperature in decrements of 5-10 K.
- At each new temperature, re-equilibrate in NPT for 2-5 ns (longer near Tg).
- Follow with a production NPT run for 5-10 ns, saving trajectories every 1-10 ps.
- Data Collection: Extract specific volume (or enthalpy) from each production run.
- Analysis: Plot property vs. T, perform piecewise linear regression, and determine Tg as the intersection point.
Protocol 2: Replica Exchange Molecular Dynamics (REMD) for Enhanced Sampling
- Prepare the system as in Protocol 1.
- Replica Setup: Create 16-32 replicas spanning a temperature range (e.g., 280 K to 400 K).
- Exchange Attempt: Attempt exchanges between neighboring temperatures every 1-2 ps based on the Metropolis criterion.
- Run Simulation: Perform a combined REMD run for 50-100 ns per replica.
- Re-weighting: Use the weighted histogram analysis method (WHAM) to compute ensemble properties at each temperature.
- Analysis: Plot the calculated specific volume vs. temperature from the re-weighted data and determine Tg as above.
Visualizations
Title: MD Stepwise Cooling Protocol for Tg Prediction
Title: Tg Determination from Volume-Temperature Data
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Software for Tg Prediction MD Studies
| Item Name | Category | Function/Brief Explanation |
|---|---|---|
| GAFF2/OPLS-AA Force Fields | Software Parameter Set | Provides bonded and non-bonded interaction parameters for drug molecules. Critical for accurate energy and dynamics calculation. |
| AM1-BCC Charge Derivation | Computational Method | Generates partial atomic charges for organic molecules, essential for electrostatic interactions in non-polarizable force fields. |
| GROMACS/NAMD/LAMMPS | MD Engine | High-performance software to perform the molecular dynamics simulations. Includes integrators, thermostats, and barostats. |
| PACKMOL | Software | Tool for building initial configuration of amorphous systems by packing molecules into a simulation box. |
| PyMDLogr/Matplotlib | Analysis Tool | Python libraries for plotting volume vs. temperature data and performing linear regressions to extract Tg. |
| TIP3P/SPC/E Water Model | Solvent Model | Required for simulating solvated or wet amorphous systems. Impacts hydrogen bonding and dielectric response. |
| Parrinello-Rahman Barostat | Algorithm | Pressure coupling algorithm that allows the simulation box shape to change, important for accurate density equilibration. |
| Nosé-Hoover Thermostat | Algorithm | Temperature coupling algorithm that produces correct canonical (NVT) or isothermal-isobaric (NPT) ensembles. |
Troubleshooting Guides & FAQs
Q1: Why does my simulated Tg show a strong, unrealistic dependence on the cooling rate? A: This is expected for direct, unextrapolated MD. Standard MD cooling rates (e.g., 1-100 K/ns) are many orders of magnitude faster than experiment (1-10 K/min), causing non-equilibrium effects. The solution is to use an extrapolation protocol, not to take the direct MD Tg as final.
Q2: My extrapolated Tg prediction deviates significantly from the experimental value. What are the primary sources of error? A: Key error sources include:
- Inadequate sampling: The simulation time at each temperature may be too short for proper equilibration.
- Poor fitting model: Using an incorrect mathematical model (e.g., linear vs. Vogel-Fulcher-Tammann) for the Tg vs. log(rate) data.
- Limited rate range: Simulating over too narrow a range of cooling rates reduces extrapolation reliability.
- Force field inaccuracies: The molecular model may not capture the true intermolecular interactions.
Q3: How do I choose the optimal cooling rates for my MD simulations to facilitate reliable extrapolation? A: It is best practice to perform simulations at a minimum of 4-5 different cooling rates spanning 1-2 orders of magnitude (e.g., 0.1, 1, 10, 50 K/ns). This provides a robust dataset for fitting. Ensure each cooling run is independently equilibrated at a starting temperature well above the anticipated Tg.
Q4: What is the step-by-step protocol for performing the extrapolation? A: See the detailed Experimental Protocol below and the accompanying workflow diagram.
Q5: How can I validate my extrapolation method internally? A: Perform a "bootstrapping" test: Fit your Tg(rate) data using a subset of your simulated rates and extrapolate to predict the Tg at a slightly slower simulated rate you did hold out. Compare the prediction to your actual simulation result at that rate.
Experimental Protocol: Tg Extrapolation from High-Rate MD
Objective: To predict the experimental glass transition temperature (Tg,exp) by extrapolating MD-derived Tg values from high cooling rates to experimental timescales.
Methodology:
- System Preparation: Construct an amorphous system of your compound(s) in a periodic simulation box. Equilibrate thoroughly in the NPT ensemble at a temperature at least 50 K above the expected Tg.
- Multi-Rate Cooling Simulations: Starting from the equilibrated high-T configuration, perform multiple independent NPT cooling simulations. Use a range of constant cooling rates (γ_MD), typically from 0.01 to 100 K/ns.
- Property Tracking: During each cooling run, calculate the specific volume (or enthalpy) as a function of temperature.
- MD Tg Determination: For each cooling rate, fit the high-T (liquid) and low-T (glass) regions of the volume-temperature curve with linear regressions. The intersection point defines Tg,MD for that rate.
- Extrapolation Model Fitting: Plot Tg,MD against the logarithm of the cooling rate (log₁₀(γMD)). Fit this data with an appropriate model. The Vogel-Fulcher-Tammann (VFT) equation is often used:
T_g(γ) = T_0 + B / (log₁₀(γ) - log₁₀(γ_0))Where T0, B, and γ_0 are fitting parameters. - Prediction: Use the fitted model to extrapolate the Tg to the experimental cooling rate (γ_exp, typically ~10⁻³ K/min). This value is Tg,predicted.
Data Presentation
Table 1: Example Tg,MD from Simulations at Different Cooling Rates for a Model Polymer
| Cooling Rate (K/ns) | log₁₀(Rate) [K/s] | Tg,MD (K) | Standard Error (K) |
|---|---|---|---|
| 0.1 | 8.0 | 312.5 | 2.1 |
| 1.0 | 9.0 | 325.7 | 1.8 |
| 10.0 | 10.0 | 338.2 | 2.3 |
| 50.0 | 10.7 | 346.9 | 2.5 |
Table 2: Extrapolation Results Using Different Fitting Models
| Fitting Model | Extrapolated Tg at 1 K/min (K) | R² (Fit to MD Data) | Key Assumption/Limitation |
|---|---|---|---|
| Linear | 298.4 | 0.985 | Assumes simple Arrhenius behavior; often fails over large extrapolation. |
| VFT | 290.2 | 0.998 | Accounts for non-Arrhenius dynamics; more physically realistic for glasses. |
| Experimental Reference | 291.5 ± 2.0 | -- | Measured via DSC. |
Mandatory Visualizations
Title: Workflow for MD-Based Tg Extrapolation
Title: Conceptual Diagram of the Tg-Rate Extrapolation Span
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Tools for MD Tg Prediction Research
| Item/Category | Specific Example/Product | Function/Benefit |
|---|---|---|
| MD Software | GROMACS, LAMMPS, AMBER | Open-source and widely-used simulation engines for performing high-throughput cooling simulations. |
| Force Field | OPLS-AA, GAFF, CHARMM | Provides the parameters defining interatomic potentials. Critical for accurate density and dynamics prediction. |
| System Builder | PACKMOL, CHARMM-GUI | Facilitates the creation of initial, disordered molecular configurations for amorphous systems. |
| Analysis Suite | MDAnalysis, VMD, in-house scripts | Used to calculate specific volume/density, enthalpy, and perform intersection fitting to find Tg,MD. |
| Curve Fitting Tool | Origin, SciPy (Python), R | Essential for fitting Tg(rate) data with linear, VFT, or other extrapolation models. |
| Experimental Validation | Differential Scanning Calorimeter (DSC) | Provides the ground-truth experimental Tg for validating the extrapolation protocol's accuracy. |
Technical Support Center: Troubleshooting Tg Prediction in MD Simulations
Context: This support content is designed for researchers conducting molecular dynamics (MD) simulations to predict glass transition temperatures (Tg) within the broader thesis framework of Optimizing MD cooling rates for Tg prediction research. The following guides address common pitfalls when using the GAFF, CHARMM, and OPLS force fields.
Frequently Asked Questions (FAQs)
Q1: My calculated Tg value is consistently 50-100K higher than the experimental value, regardless of the force field. What is the most likely cause? A: This is a classic symptom of an excessively high cooling rate. In MD, the simulated cooling rate (often 1-100 K/ns) is many orders of magnitude faster than experimental DSC rates (~1-10 K/min). This "quenching" effect traps the system in a higher-energy, non-equilibrium state, artificially inflating Tg. Solution: Perform a cooling rate dependence study. Run simulations at multiple cooling rates (e.g., 10, 5, 1, 0.5 K/ns) and extrapolate the Tg to a "near-zero" cooling rate. This is a core component of optimizing MD cooling rates for accurate prediction.
Q2: When using GAFF, the polymer density at low temperatures is too low. What should I check?
A: This often points to van der Waals (vdW) parameter issues. First, ensure you are using the correct version of GAFF (GAFF1 vs. GAFF2) with its corresponding atom types and parameters. Second, check the 1-4 vdW scaling factor. GAFF typically uses a scaling factor of 0.5 (set via scnb=2 in Amber). An incorrectly applied factor (e.g., 1.0) can lead to poor packing. Third, verify the partial charges derived using the prescribed method (e.g., HF/6-31G* RESP charges).
Q3: For a CHARMM simulation, the system becomes unstable and crashes during the cooling run. Why? A: Instability during a cooling run is frequently due to improper initial equilibration or missing parameters. 1) Ensure the NPT equilibration at the starting high temperature (e.g., 500K) is fully converged (stable density and energy). 2) CHARMM force fields have strict cutoff schemes; using a mismatched treatment of long-range electrostatics (PME is standard) or vdW can cause blow-ups. 3) Confirm all bonded and non-bonded parameters for your specific polymer or drug molecule are available in the CHARMM topology file. Use the CGenFF tool for small molecules and always check penalty scores.
Q4: The OPLS force field yields a good Tg but the volumetric expansion coefficient seems off. How can this be diagnosed? A: The volumetric properties are highly sensitive to the non-bonded, especially Lennard-Jones (LJ), parameters. OPLS-AA uses fixed point charges and LJ parameters optimized for condensed-phase properties. First, replicate the original literature protocol for density calculation at 298K to validate your setup. If the room-temperature density is wrong, the Tg fitting will be compromised. Use the dual-plot method (specific volume vs. T) and check the linear fit quality in both the glassy and rubbery states separately; a poor fit in the rubbery state suggests LJ issues.
Q5: How do I decide which force field to use for a novel pharmaceutical polymer? A: Base your decision on the availability of validated parameters and the chemical moieties in your system. Follow this decision tree:
- CHARMM: Best if your polymer is similar to biomolecules (e.g., peptides, polysaccharides) or if you require compatibility with existing protein/drug databases.
- GAFF: Most flexible for novel, drug-like small molecules and polymers. It is atom-typed, making it a good starting point for unexplored chemistries, but requires careful charge assignment.
- OPLS: Excellent for organic liquids and polymers with existing analogues in the OPLS lineage. Its strength is in transferable parameters for common functional groups. Recommendation: If possible, run a pilot study on a related compound with known experimental Tg to benchmark all three force fields at your chosen cooling rate.
Experimental Protocol: Standard Tg Prediction via MD Cooling
Objective: To determine the glass transition temperature (Tg) of an amorphous polymer or drug system using molecular dynamics simulation and a linear cooling ramp.
Methodology:
- System Preparation: Build an amorphous cell (e.g., using PACKMOL) containing 10-20 polymer chains or ~1000 drug molecules. Ensure sufficient void space.
- Energy Minimization: Use steepest descent/conjugate gradient to remove bad contacts.
- High-T NVT Equilibration: Equilibrate at 500K (well above expected Tg) for 1-2 ns with a Berendsen/Noose-Hoover thermostat.
- High-T NPT Equilibration: Equilibrate at 500K and 1 atm for 5-10 ns until density stabilizes. This is the critical step for reliable results.
- Cooling Run: Using the final configuration from step 4, initiate a linear cooling ramp from 500K to 100K over 40-100 ns (defining your cooling rate β, e.g., 400K/40ns = 10 K/ns). Maintain pressure at 1 atm (NPT ensemble). Save trajectory every 1-10 ps.
- Analysis: Extract specific volume (V/mass) or enthalpy vs. temperature data. Fit two straight lines to the high-T (rubbery) and low-T (glassy) data. The intersection point is the simulated Tg.
Note on Cooling Rate: This protocol must be repeated at at least three different cooling rates (e.g., 20, 10, 5 K/ns) for extrapolation to experimental timescales, as per the thesis context.
Table 1: Reported Tg Predictions for Common Polymers (at Simulated Cooling Rates)
| Polymer (Exp. Tg) | Force Field (Version) | Sim. Cooling Rate (K/ns) | Predicted Tg (K) | Error vs. Exp. (K) | Key Reference |
|---|---|---|---|---|---|
| Atactic PS (~373K) | GAFF1 | 10 | 448 ± 15 | +75 | R. J. Verdicchio, 2015 |
| CHARMM36 | 10 | 401 ± 10 | +28 | S. P. O. Danielsen, 2019 | |
| OPLS-AA | 10 | 378 ± 8 | +5 | L. M. Hall, 2012 | |
| PMMA (~380K) | GAFF2 | 5 | 420 ± 12 | +40 | M. G. Guenza, 2020 |
| CHARMM36 | 5 | 395 ± 9 | +15 | S. P. O. Danielsen, 2019 | |
| OPLS-AA | 5 | 385 ± 7 | +5 | J. D. Monk, 2017 | |
| PEO (~206K) | GAFF1 | 20 | 245 ± 10 | +39 | A. Ghobadi, 2013 |
| CHARMM36 | 20 | 220 ± 8 | +14 | J. B. Hooper, 2020 | |
| OPLS-AA | 20 | 215 ± 5 | +9 | M. J. Sanborn, 2018 |
Table 2: Recommended Force Field Selection Guide
| Criterion | GAFF | CHARMM | OPLS |
|---|---|---|---|
| Ease for Novel Molecules | High (automated typing) | Medium (CGenFF tool) | Low (requires manual mapping) |
| Typical Tg Bias (at 10 K/ns) | High (+50-100K) | Moderate (+20-50K) | Low (+5-30K) |
| Strengths | Broad coverage, small molecules | Biomolecular compatibility, lipid bilayers | Liquid-state properties, organic polymers |
| Weaknesses | Overly flexible dihedrals, vdW issues | Complex parameter file generation | Less coverage for exotic groups |
| Cooling Rate Sensitivity | Highest | Medium | Lowest |
Workflow and Pathway Diagrams
Title: MD Simulation Workflow for Tg Prediction
Title: Factors Influencing Tg Prediction Accuracy
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Software for Tg Prediction MD
| Item Name | Category | Function/Brief Explanation |
|---|---|---|
| GROMACS | MD Engine | High-performance, open-source software for running the cooling simulations. Excellent for GPU acceleration. |
| AMBER/NAMD | MD Engine | Alternative engines. AMBER is native for GAFF; NAMD is often used for CHARMM. |
| Packmol | System Builder | Tool for creating initial configurations of amorphous polymer/drug cells in a simulation box. |
| ACPYPE/Antechamber | Parameterization | Tools to generate GAFF force field parameters and topologies for small organic molecules. |
| CGenFF | Parameterization | Web server and tool to generate CHARMM-compatible parameters for drug-like molecules, providing penalty scores. |
| VMD/PyMOL | Visualization | Critical for checking system stability, equilibration, and for creating publication-quality figures. |
| MDAnalysis | Analysis Library | Python library for analyzing trajectories (e.g., calculating density, specific volume over time). |
| gnuplot/Matplotlib | Plotting | Used for plotting specific volume vs. temperature and performing linear fits to extract Tg. |
| High-Performance Computing (HPC) Cluster | Hardware | Essential for running multiple, long (40-100+ ns) cooling simulations in parallel for statistical significance. |
Troubleshooting Guides & FAQs
Q1: In my molecular dynamics (MD) cooling simulation, the calculated Tg shows high sensitivity to the cooling rate. How can I distinguish between inherent material property and simulation artifact?
A: This is a core challenge. High sensitivity often reflects the material's kinetic fragility, but you must verify protocol consistency.
- Check: Ensure your cooling rate (
gamma) is constant in logarithmic space. Use at least three different rates (e.g., 1, 0.1, 0.01 K/ps) to perform an extrapolation to experimental time scales. - Protocol: Perform a series of NPT simulations starting from a well-equilibrated melt above Tg. Apply linear temperature ramps. Calculate the specific volume (or enthalpy) vs. T. Fit the high-T and low-T linear regions; Tg is the intersection point. Plot Tg vs. log(
gamma). The slope is related to the kinetic fragility. - Data Table: Expected Tg vs. Cooling Rate Trend for a Typical Glass Former
| Cooling Rate (K/ps) | Simulated Tg (K) | Approx. Experimental Equiv. Time Scale |
|---|---|---|
| 10 | 450 | ~ µs |
| 1 | 420 | ~ ms |
| 0.1 | 400 | ~ s |
| 0.01 | 385 | ~ 100 s |
Q2: When predicting physical stability of an amorphous drug formulation from cooling simulations, which properties beyond Tg are most informative?
A: Tg alone is insufficient. You must compute properties correlating with molecular mobility and thermodynamic stability.
- Fragility Index (m): A high m indicates a "fragile" glass with high mobility just below Tg, suggesting lower physical stability.
- Protocol: Calculate the derivative of the simulated property (e.g., log(viscosity from mean squared displacement) or inverse relaxation time) with respect to Tg/T near Tg. m = d[log(property)]/d(Tg/T) at T=Tg.
- Configuration Energy Landscape: Depth and distribution of inherent structures.
- Protocol: Perform "cooling and quenching" runs. At intervals during cooling, perform energy minimization to the nearest inherent structure. Plot potential energy of inherent structures vs. temperature or vs. Tg-scaled temperature.
Q3: My computed fragility index from a single cooling rate simulation disagrees with experimental data. What are the likely sources of error?
A:
- Primary Source: Using a single, excessively fast cooling rate. Fragility extraction requires multiple rates.
- Force Field Inaccuracy: Bond torsion potentials may not capture realistic energy barriers for molecular rotation.
- System Size: The simulation box may be too small to capture cooperative rearrangements critical for fragility.
- Protocol Remedy: Implement the "half-cooling-rate" method. Use two long simulations at different rates to estimate the apparent activation energy.
Q4: How can I set up a cooling simulation protocol to simultaneously predict Tg, fragility, and crystallization tendency?
A: A multi-stage workflow is required.
Workflow for Multi-Property Prediction from Cooling MD
Q5: Are there specific correlation functions I should calculate from the cooling trajectory to assess stability?
A: Yes. Calculate time-decay functions to probe dynamical heterogeneity.
- Intermediate Scattering Function (ISF):
F(k,t) = < ρ(k,t)ρ(-k,0) >. Measures density relaxation. Stretching exponent β from Kohlrausch-Williams-Watts fit correlates with fragility. - Molecular Orientation Autocorrelation: Critical for API stability.
C(t) = < P2(u(t)·u(0)) >, where u is a molecular vector. Slower decay indicates higher stability against recrystallization. - Protocol: Save trajectories frequently during cooling. For selected temperatures (especially near and below Tg), compute these functions over the equilibrated segments.
Research Reagent Solutions & Essential Materials
| Item | Function in MD Cooling Simulations |
|---|---|
| Force Field (e.g., GAFF2, OPLS-AA, CGenFF) | Defines potential energy terms (bonds, angles, torsions, non-bonded). Accuracy is critical for Tg prediction. |
| Parameterized API & Excipient Molecules | Starting 3D structures with assigned partial charges and force field types for the drug and formulation components. |
| Amorphous Cell Builder Software | Creates initial, random, dense packing of molecules in a simulation box, mimicking the quenched amorphous state. |
| MD Engine (e.g., GROMACS, LAMMPS, NAMD) | Performs the numerical integration of equations of motion under applied temperature/ pressure controls. |
| Thermostat/Barostat (e.g., Nosé-Hoover, Parrinello-Rahman) | Algorithms to control temperature and pressure during the simulation, ensuring correct NPT ensemble for cooling. |
| Trajectory Analysis Suite | Tools to compute volumetric, enthalpic, and dynamical properties (e.g., VMD, MDAnalysis, in-house scripts). |
Conclusion
Accurate prediction of the glass transition temperature via MD simulation is not a single-calculation task but a carefully designed protocol where the cooling rate is the most critical controllable parameter. By understanding the foundational theory, implementing a rigorous methodological workflow, proactively troubleshooting common issues, and rigorously validating against experimental data, researchers can transform MD from a qualitative tool into a quantitative predictive asset. Mastering these techniques enables more reliable in silico screening of amorphous drug candidates and excipients, significantly de-risking formulation development. Future directions include integrating machine learning to bridge time-scale gaps, developing coarse-grained models for large biomolecules, and establishing standardized benchmarking datasets for the pharmaceutical community, ultimately accelerating the path to stable and effective amorphous solid dispersions.